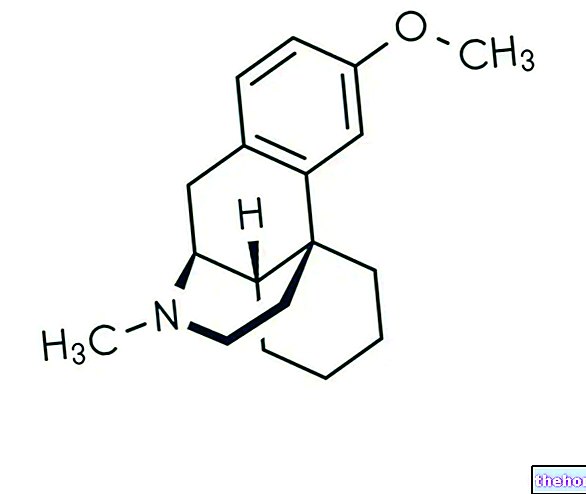
Aktive ingredienser: Dimethylfumarat (dimethylis fumaras)
Tecfidera 120 mg gastro-resistente hårde kapsler
Tecfidera 240 mg gastro-resistente hårde kapsler
Hvorfor bruges Tecfidera? Hvad er det for?
Hvad er Tecfidera
Tecfidera er et lægemiddel, der indeholder det aktive stof dimethylfumarat.
Hvad er Tecfidera
Tecfidera bruges til behandling af recidiverende remitterende multipel sklerose (MS).
Multipel sklerose (MS) er en kronisk sygdom, der påvirker centralnervesystemet (CNS), herunder hjernen og rygmarven. Tilbagefaldsgivende MS er kendetegnet ved gentagne angreb (tilbagefald) af symptomer, der påvirker nervesystemet. Symptomerne varierer fra patient til patient, men inkluderer typisk gangbesvær, en følelse af ubalance og synsbesvær. Disse symptomer kan forsvinde fuldstændigt, når tilbagefaldet forsvinder, men der kan stadig være nogle problemer.
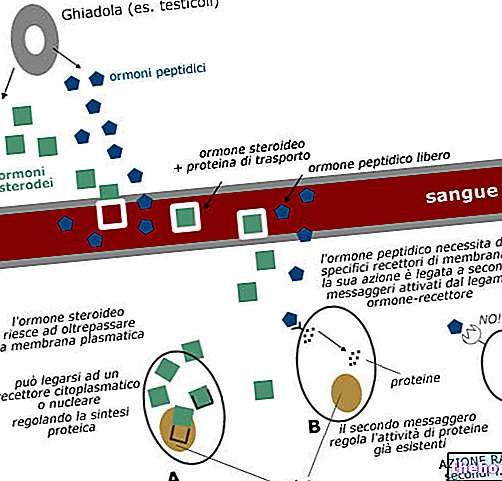
Sådan fungerer Tecfidera
Tecfidera ser ud til at virke ved at forhindre kroppens forsvar i at beskadige hjernen og rygmarven. Dette kan også hjælpe med at forsinke fremtidig forværring af multipel sklerose.
Kontraindikationer Når Tecfidera ikke bør bruges
Tag ikke Tecfidera:
- hvis du er allergisk over for dimethylfumarat eller et af de øvrige indholdsstoffer i denne medicin
Forholdsregler ved brug Hvad du skal vide, før du tager Tecfidera
Tecfidera kan påvirke antallet af hvide blodlegemer i blod, nyrer og lever. Inden du begynder at tage Tecfidera, vil din læge foretage en blodprøve for at tælle antallet af dine hvide blodlegemer og kontrollere, at dine nyrer og lever fungerer korrekt. Lægen vil regelmæssigt foretage tests under behandlingen. Hvis dit antal hvide blodlegemer falder under behandlingen, kan din læge overveje at stoppe behandlingen.
Tal med din læge, før du tager Tecfidera, hvis du har:
- alvorlig nyresygdom
- alvorlig leversygdom
- mave eller tarmsygdom
- en "alvorlig infektion (såsom lungebetændelse)
Børn og unge
Tecfidera bør ikke bruges til børn og unge under 18 år. Sikkerheden og effekten af Tecfidera er ikke kendt i denne aldersgruppe.
Interaktioner Hvilke lægemidler eller fødevarer kan ændre effekten af Tecfidera
Fortæl det til din læge eller apotek, hvis du tager anden medicin eller har brugt det for nylig.
- Lægemidler, der indeholder fumarsyreestere (fumarater), der bruges til behandling af psoriasis.
- Medicin, der påvirker kroppens immunsystem, herunder andre lægemidler, der bruges til behandling af multipel sklerose, såsom fingolimod, natalizumab eller mitoxantron, eller nogle almindeligt anvendte kræftbehandlinger.
- Medicin, der påvirker nyrerne, herunder nogle antibiotika (bruges til behandling af infektioner), diuretika, nogle typer smertestillende midler (såsom ibuprofen og andre lignende antiinflammatoriske lægemidler og medicin købt uden recept fra en læge) og medicin, der indeholder litium .
- Vaccinationer, der gives, mens du tager Tecfidera, kan være mindre effektive end normalt.Tagning af Tecfidera med visse typer vacciner (levende vacciner) kan få dig til at blive inficeret og bør derfor undgås.
Brug af Tecfidera sammen med mad, drikke og alkohol
Indtagelse af alkoholholdige drikkevarer (mere end 30% alkohol i volumen, f.eks. Likører) større end en lille mængde (mere end 50 ml) bør undgås inden for en "time efter indtagelse af Tecfidera, fordi" alkoholen kan interagere med denne medicin. Dette kan forårsage betændelse i maven (gastritis), især hos mennesker, der allerede er tilbøjelige til gastritis.
Advarsler Det er vigtigt at vide, at:
Graviditet og amning
Hvis du er gravid, tror at du er gravid eller planlægger at blive gravid, skal du spørge din læge eller apotek til råds, før du tager denne medicin.
Graviditet
Brug ikke Tecfidera, hvis du er gravid, medmindre du har diskuteret dette med din læge
Fodringstid
Det vides ikke, om ingredienserne i Tecfidera passerer i modermælk. Tecfidera må ikke bruges under amning. Din læge vil hjælpe dig med at beslutte, om du skal stoppe med at amme eller behandle med Tecfidera. Dette vejer fordelen ved amning for din baby kontra fordelene ved behandling for hende.
Kørsel og brug af maskiner
Effekten af Tecfidera på evnen til at føre motorkøretøj eller betjene maskiner kendes ikke. Din læge vil fortælle dig, om din sygdom giver dig mulighed for at køre bil og betjene maskiner sikkert.
Dosis, metode og administrationstidspunkt Sådan bruges Tecfidera: Dosering
Tag altid denne medicin nøjagtigt efter lægens anvisning. Hvis du er i tvivl, skal du kontakte din læge eller apotek.
Indledende dosis
120 mg to gange dagligt.
Tag denne startdosis i de første 7 dage, og tag derefter den normale dosis
Regelmæssig dosis
240 mg to gange dagligt.
Synk hver kapsel hel, med lidt vand. Du må ikke splitte, knuse, opløse, suge eller tygge kapslen, da dette kan øge nogle uønskede virkninger.
Tag Tecfidera sammen med mad - det kan hjælpe med at reducere nogle mere almindelige bivirkninger (angivet i afsnit 4).
Overdosering Hvad skal jeg gøre, hvis du har taget for meget Tecfidera
Hvis du har taget for mange Tecfidera
Hvis du har taget for mange kapsler, skal du straks kontakte din læge.
Hvis du har glemt at tage Tecfidera
Hvis du glemmer eller glemmer en dosis, må du ikke tage en dobbeltdosis som erstatning for en glemt dosis.
Du kan tage den glemte dosis, hvis du lader mindst 4 timer gå mellem doserne. Ellers skal du vente til den næste planlagte dosis.
Spørg din læge eller apotek, hvis du har yderligere spørgsmål om brugen af dette lægemiddel.
Bivirkninger Hvad er bivirkningerne af Tecfidera
Ligesom al anden medicin kan denne medicin forårsage bivirkninger, men ikke alle får bivirkninger.
Alvorlige bivirkninger
Meget lave niveauer af lymfocytter - Antallet af lymfocytter (en type hvide blodlegemer) kan falde over en lang periode. Vedligeholdelse af lave niveauer af hvide blodlegemer over en længere periode kan øge risikoen for infektioner, herunder risikoen for en sjælden hjerneinfektion kaldet progressiv multifokal leukoencefalopati (PML). Symptomer på PML kan ligne symptomer på et MS -tilbagefald. Symptomer kan omfatte begyndelsen eller forværringen af svaghed på den ene side af kroppen (hemiparesis); dårlig koordination; ændringer i syn, tænkning eller hukommelse; eller forvirring eller personlighedsændringer, der varer mere end et par dage.
- Hvis du får nogle af disse symptomer, skal du straks kontakte din læge.
Allergiske reaktioner - disse er ikke almindelige og kan forekomme hos op til 1 ud af 100 mennesker
Rødme i ansigtet eller kroppen (rødme) er en meget almindelig bivirkning (kan forekomme hos mere end 1 ud af 10 personer), men hvis du har rødme og har et af disse tegn:
- hævelse af ansigt, læber, mund eller tunge
- hvæsen, vejrtrækningsbesvær eller åndenød
Stop med at tage Tecfidera og kontakt straks en læge.
Meget almindelige bivirkninger
Disse kan påvirke mere end 1 ud af 10 personer:
- rødme i ansigtet eller kroppen, varme, varme, brændende eller kløe (rødme)
- løs afføring (diarré)
- følelse af forestående opkastning (kvalme)
- mavesmerter eller mavekramper
At tage medicinen med mad kan hjælpe med at reducere de bivirkninger, der er nævnt ovenfor.
Tilstedeværelsen af stoffer kaldet ketoner, som naturligt produceres af kroppen, afsløres meget almindeligt i urinprøver, mens de tager Tecfidera.
Tal med din læge for at få oplysninger om, hvordan du håndterer disse bivirkninger. Din læge kan reducere din dosis. Reducer ikke dosis, medmindre din læge fortæller dig det.
Almindelige bivirkninger
Disse kan påvirke op til 1 ud af 10 personer:
- betændelse i tarmens foring (gastroenteritis)
- Han trak sig tilbage
- fordøjelsesbesvær (dyspepsi)
- betændelse i maveslimhinden (gastritis)
- mave -tarmsygdom
- brændende sensation
- hedeture, følelse af varme
- kløende (kløende) hud
- udslæt
- lyserøde eller røde pletter på huden (erytem)
Almindelige bivirkninger, som kan dukke op ved blod- eller urintest
- lavt niveau af hvide blodlegemer (lymfocytopeni, leukopeni) i blodet. Reduktionen i antallet af hvide blodlegemer kan indikere, at din krop er mindre i stand til at bekæmpe en "infektion. Hvis du har en" alvorlig infektion (f.eks. Lungebetændelse), skal du straks kontakte din læge.
- protein (albumin) i urinen
- forhøjede niveauer af leverenzymer (alaninaminotransferase, ALAT og aspartataminotransferase, ASAT) i blodet
Indberetning af bivirkninger
Tal med din læge eller apotek, hvis du får bivirkninger, herunder mulige bivirkninger, som ikke er nævnt i denne indlægsseddel. Du kan også indberette bivirkninger direkte via det nationale rapporteringssystem anført i tillæg V. Ved at rapportere bivirkninger kan du hjælpe med at give mere information om sikkerheden ved dette lægemiddel.
Udløb og opbevaring
Opbevar denne medicin utilgængeligt for børn.
Brug ikke dette lægemiddel efter den udløbsdato, der står på kartonen efter "EXP". Udløbsdatoen refererer til den sidste dag i den pågældende måned.
Må ikke opbevares over 30 ° C.
Opbevares i den originale emballage for at beskytte medicinen mod lys.
Smid ikke medicin via spildevand eller husholdningsaffald. Spørg din apotek om, hvordan du skal smide medicin, du ikke længere bruger. Dette vil hjælpe med at beskytte miljøet.
Indeholder Tecfidera
Den aktive ingrediens er dimethylfumarat. Tecfidera 120 mg: Hver kapsel indeholder 120 mg dimethylfumarat.
Tecfidera 240 mg: Hver kapsel indeholder 240 mg dimethylfumarat.
Øvrige indholdsstoffer er mikrokrystallinsk cellulose, croscarmellosenatrium, talkum, kolloid vandfri silica, magnesiumstearat, triethylcitrat, methacrylsyre-methylmethacrylatcopolymer (1: 1), methacrylsyre-ethylacrylatcopolymer (1: 1) dispersion 30%, simethicon , natriumlaurylsulfat, polysorbat 80, gelatine, titandioxid (E171), Brilliant Blue FCF (E133), gult jernoxid (E172), shellak, kaliumhydroxid og sort jernoxid (E172).
Beskrivelse af hvordan Tecfidera ser ud og pakningens indhold
Tecfidera 120 mg gastroresistente hårde kapsler er grønne og hvide og præget med 'BG-12 120 mg' og fås i pakninger indeholdende 14 kapsler.
Tecfidera 240 mg hårde gastro-resistente kapsler er grønne og præget med 'BG-12 240 mg' og fås i pakninger indeholdende 56 eller 168 kapsler.
Ikke alle pakningsstørrelser er nødvendigvis markedsført.
Indlægsseddel: AIFA (Italian Medicines Agency). Indhold offentliggjort i januar 2016. De foreliggende oplysninger er muligvis ikke opdaterede.
For at få adgang til den mest opdaterede version er det tilrådeligt at få adgang til webstedet AIFA (Italian Medicines Agency). Ansvarsfraskrivelse og nyttige oplysninger.
01.0 LÆGEMIDLETS NAVN
TECFIDERA 120 - 240 MG HARDE GASTRORESISTENTE KAPPELER.
02.0 KVALITATIV OG KVANTITATIV SAMMENSÆTNING
Tecfidera 120 mg kapsel
Hver kapsel indeholder 120 mg dimethylfumarat.
Tecfidera 240 mg kapsel
Hver kapsel indeholder 240 mg dimethylfumarat
Den fulde liste over hjælpestoffer findes i afsnit 6.1.
03.0 LÆGEMIDDELFORM
Hård gastro-resistent kapsel
Tecfidera 120 mg kapsel
Grøn og hvid gastro-resistent hård kapsel, præget med "BG-12 120 mg".
Tecfidera 240 mg kapsel
Grøn kapsel, hård, gastro-resistent, præget med "BG-12 240 mg"
04.0 KLINISKE OPLYSNINGER
04.1 Terapeutiske indikationer
Tecfidera er indiceret til behandling af voksne patienter med recidiverende remitterende multipel sklerose (se pkt.5.1 for vigtig information om de populationer, for hvilke der er påvist effekt).
04.2 Dosering og indgivelsesmåde
Behandlingen bør påbegyndes under opsyn af en læge med erfaring i behandling af sygdommen.
Dosering
Startdosis er 120 mg to gange dagligt. Efter 7 dage øges dosis til den anbefalede dosis på 240 mg to gange dagligt.
Midlertidig dosisreduktion til 120 mg to gange dagligt kan reducere begyndelsen af rødme og gastrointestinale bivirkninger. Inden for 1 måned skal den anbefalede dosis på 240 mg to gange dagligt genoptages.
Tecfidera skal tages sammen med mad (se afsnit 5.2). Indtagelse af Tecfidera sammen med mad kan forbedre tolerabiliteten hos de patienter, der kan være tilbøjelige til rødme eller gastrointestinale bivirkninger (se pkt. 4.4, 4.5 og 4.8).
Ældre borgere
Kliniske undersøgelser af Tecfidera omfattede et begrænset antal patienter i alderen 55 år og derover og inkluderede ikke et tilstrækkeligt antal patienter i alderen 65 år og derover til at afgøre, om de reagerer anderledes end yngre patienter (se pkt. 5.2). Baseret på virkningsmekanismen for det aktive stof er der ingen teoretisk grund til, at dosisjusteringer er nødvendige hos ældre.
Nedsat nyre- og leverfunktion
Tecfidera er ikke undersøgt hos patienter med nedsat nyre- eller leverfunktion. Baseret på kliniske farmakologiske undersøgelser er der ingen dosisjusteringer påkrævet (se pkt. 5.2). Der skal udvises forsigtighed ved behandling af patienter med svært nedsat nyrefunktion eller svært nedsat leverfunktion (se pkt. 4.4).
Pædiatrisk population
Sikkerheden og effekten af Tecfidera hos børn og unge i alderen 10 til 18 år er endnu ikke fastslået. Der er ingen tilgængelige data. Der er ingen relevant brug af Tecfidera til børn under 10 år til "Tilbagevendende remitterende multipel sklerose -indikation.
Indgivelsesmåde
Til oral brug.
Kapslen eller dens indhold må ikke knuses, deles, opløses, suges eller tygges, da mikro-tabletternes enteriske belægning forhindrer irriterende virkninger på tarmen.
04.3 Kontraindikationer
Overfølsomhed over for det aktive stof eller over for et eller flere af hjælpestofferne anført i pkt.6.1.
04.4 Særlige advarsler og passende forholdsregler ved brug
Blodprøver / laboratorieanalyser
Laboratorieændringer i nyre- og leverfunktion er blevet observeret i kliniske undersøgelser hos personer behandlet med Tecfidera (se pkt. 4.8). De kliniske konsekvenser af disse ændringer er ukendte. Vurdering af nyrefunktion (f.eks. Kreatinin, urinstofværdier i blodet og urinalyse) og leverfunktion (f.eks. Alaninaminotransferase, ALAT og aspartataminotransferase (ASAT) anbefales inden behandlingsstart, efter 3 og 6 måneders behandling og derefter hver 6-12 måned, som klinisk angivet.
Patienter behandlet med Tecfidera kan udvikle alvorlig og langvarig lymfopeni (se pkt. 4.8). Tecfidera er ikke undersøgt hos patienter med eksisterende lavt antal lymfocytter, og der skal udvises forsigtighed, når disse patienter behandles. Inden behandlingen påbegyndes med Tecfidera, bør der foretages et opdateret fuldstændigt blodtal, inklusive lymfocytter. Hvis der findes lymfocyttal under det normale område, bør der foretages en omhyggelig evaluering af mulige årsager, før behandlingen med Tecfidera påbegyndes.
Efter behandlingsstart bør et fuldstændigt blodtal, inklusive lymfocytter, evalueres hver 3. måned. Hos patienter med lymfocyttal
Lymfocyttal bør overvåges, indtil de genopretter. Efter helbredelse og i mangel af alternative behandlingsmuligheder, bør beslutninger om, hvorvidt Tecfidera skal genstartes eller ikke seponeres, baseres på klinisk vurdering.
Magnetic resonance imaging (MRI)
Inden behandling påbegyndes med Tecfidera, bør en baseline MR (normalt inden for 3 måneder) være tilgængelig til brug som reference. Behovet for yderligere magnetiske resonansbilleder (MRI) undersøgelser bør vurderes i overensstemmelse med nationale og lokale anbefalinger. Magnetisk resonansbilleddannelse (MR -billeddannelse) kan betragtes som en del af øget opmærksomhed hos patienter, der anses for at have øget risiko for PML. Hvis der er klinisk mistanke om PML, bør der straks foretages en MR -scanning til diagnostiske formål.
Progressiv multifokal leukoencefalopati (PML)
Med Tecfidera og andre produkter, der indeholder fumarat, har der været tilfælde af PML i "indstilling af alvorlig og langvarig lymfopeni. PML er en" opportunistisk infektion forårsaget af John Cunninghams virus (JCV), som kan være dødelig eller resultere i alvorlig handicap.PML kan kun forekomme i nærvær af en JCV-infektion. Ved test af JCV skal det tages i betragtning, at lymfopeniens indflydelse på nøjagtigheden af anti-JCV-antistoftesten ikke er undersøgt hos behandlede patienter. Det er også nødvendigt at husk på, at en negativ test for tilstedeværelsen af anti-JCV-antistoffer (i nærvær af normale lymfocyttal) ikke udelukker muligheden for JCV-infektion i fremtiden.
Tidligere behandling med immunsuppressive eller immunmodulerende behandlinger
Der er ikke udført undersøgelser for at evaluere effekten og sikkerheden af Tecfidera hos patienter, der skifter fra andre sygdomsmodificerende terapier til Tecfidera.Bidraget fra tidligere immunsuppressive behandlinger i udviklingen af PML hos patienter behandlet med Tecfidera er ukendt.Når patienter skifter fra en anden sygdom -modificerende behandling af Tecfidera, halveringstiden og virkningsmåden for den anden terapi skal tages i betragtning for at undgå en additiv effekt på immunsystemet og samtidig reducere risikoen for MS-reaktivering.
Det anbefales at foretage et fuldstændigt blodtal før behandling med Tecfidera påbegyndes og med jævne mellemrum under behandlingen (se Blodprøver / laboratorieanalyser på).
Generelt kan behandling med Tecfidera startes umiddelbart efter afbrydelse af interferon eller glatirameracetat.
Alvorligt nedsat nyre- og leverfunktion
Tecfidera er ikke undersøgt hos patienter med svært nedsat nyrefunktion eller svært nedsat leverfunktion, og derfor bør der udvises forsigtighed hos disse patienter (se pkt.4.2).
Alvorlig aktiv mave -tarmsygdom
Tecfidera er ikke undersøgt hos patienter med alvorlig aktiv mave -tarmsygdom, og derfor bør der udvises forsigtighed hos disse patienter.
Rødme (rødme)
I kliniske undersøgelser oplevede 34% af patienterne behandlet med Tecfidera rødme. Hos de fleste patienter med rødme var dette mildt eller moderat.
I kliniske undersøgelser oplevede 3 ud af i alt 2.560 patienter, der blev behandlet med Tecfidera, alvorlige symptomer på rødme, muligvis forårsaget af overfølsomhed eller anafylaktoide reaktioner. Disse hændelser var ikke livstruende, men krævede hospitalsindlæggelse. Læger og patienter bør være opmærksom på denne mulighed i tilfælde af alvorlige skylningsreaktioner (se pkt. 4.2, 4.5 og 4.8).
Infektioner
I fase III placebokontrollerede undersøgelser var forekomsten af infektioner (60% versus 58%) og alvorlige infektioner (2% versus 2%) ens hos patienter behandlet med henholdsvis Tecfidera eller placebo. Det blev ikke observeret. En øget forekomst alvorlige infektioner hos patienter med stabiliserende lymfocyttal (se pkt.4.8). Det gennemsnitlige lymfocyttal forblev inden for normale grænser. Lymfocyttal
Hvis behandlingen fortsættes i nærvær af alvorlig og langvarig lymfopeni, kan risikoen for opportunistiske infektioner, herunder lymfopeni, ikke udelukkes.progressiv multifokal eukoencephalopati (PML) (se underafsnittet om PML ovenfor for yderligere detaljer).
Hvis en patient udvikler en alvorlig infektion, skal afbrydelse af Tecfidera overvejes, og fordele og risici skal vurderes før behandling genstartes Patienter på Tecfidera bør rådes til at rapportere symptomer på infektioner til læge Patienter med alvorlige infektioner bør ikke starte Tecfidera, før infektionen ) er løst.
04.5 Interaktioner med andre lægemidler og andre former for interaktion
Tecfidera er ikke undersøgt i kombination med antineoplastiske eller immunsuppressive behandlinger, og derfor bør der udvises forsigtighed under samtidig administration. I kliniske forsøg med multipel sklerose var samtidig behandling af tilbagefald med et kort forløb af intravenøse kortikosteroider ikke forbundet med en klinisk relevant stigning i infektionen.
Vaccination under Tecfidera -behandling er ikke blevet evalueret. Det vides ikke, om behandling med Tecfidera vil reducere effekten af nogle vacciner. Levende vacciner kan have en øget risiko for klinisk infektion og bør ikke gives til patienter behandlet med Tecfidera, medmindre denne potentielle risiko i særlige tilfælde anses for mindre vigtig end risikoen for ikke-vaccination for den enkelte.
Under behandling med Tecfidera bør samtidig brug af andre fumarsyrederivater (topiske eller systemiske) undgås.
Hos mennesker metaboliseres dimethylfumarat i vid udstrækning af esteraser, før det når den systemiske cirkulation, og yderligere metabolisme sker gennem tricarboxylsyre -cyklussen uden involvering af cytochrom P450 (CYP) -systemet. Ingen potentielle risici ved lægemiddelinteraktioner blev identificeret fra undersøgelserne in vitro inhibering og induktion af CYP, fra en undersøgelse af p-glycoproteiner eller fra proteinbindende undersøgelser af dimethylfumarat og monomethylfumarat (en primær metabolit af dimethylfumarat).
Lægemidler, der almindeligvis anvendes til patienter med multipel sklerose, såsom intramuskulært administreret interferon beta-1a og glatirameracetat, er klinisk testet for potentielle interaktioner med dimethylfumarat og har ikke ændret dimethylfumaratets farmakokinetiske profil.
I en undersøgelse foretaget hos raske frivillige ændrede administration af 325 mg (eller ækvivalent) ikke-enterisk coated acetylsalicylsyre 30 minutter før Tecfidera i løbet af 4 dages dosering ikke Tecfideras farmakokinetiske profil og reducerede "Begyndelse og sværhedsgrad af skylning. Langvarig brug af acetylsalicylsyre anbefales imidlertid ikke til behandling af rødme. De potentielle risici forbundet med behandling med acetylsalicylsyre bør overvejes før samtidig administration. Med Tecfidera (se pkt. 4.2, 4.4 og 4.8).
Samtidig behandling med nefrotoksiske lægemidler (såsom aminoglycosider, diuretika, NSAID eller lithium) kan øge potentielle nyrebivirkninger (f.eks. Proteinuri) hos patienter behandlet med Tecfidera (se pkt.4.8).
Forbrug af moderate mængder alkohol ændrede ikke eksponeringen for Tecfidera og var ikke forbundet med en stigning i bivirkninger.Forbrug af store mængder alkoholholdige drikkevarer (mere end 30% alkohol i volumen) kan resultere i en stigning i opløsningshastigheden Tecfidera og kan derfor øge hyppigheden af gastrointestinale bivirkninger.
Undersøgelser in vitro af CYP -induktion viste ikke en interaktion mellem Tecfidera og orale præventionsmidler. Der er ikke udført undersøgelser in vivo om interaktion med orale præventionsmidler Selvom der ikke forventes nogen interaktion, bør ikke-hormonelle præventionsmidler med Tecfidera overvejes (se pkt. 4.6).
Pædiatrisk population
Interaktionsundersøgelser er kun blevet udført hos voksne.
04.6 Graviditet og amning
Graviditet
Ingen data eller begrænset mængde data er tilgængelige om brug af dimethylfumarat til gravide Dyrestudier har vist reproduktionstoksicitet (se pkt. 5.3). Tecfidera anbefales ikke under graviditet og til kvinder i den fertile alder. Ikke at anvende passende præventionsmidler ( se afsnit 4.5) Tecfidera bør kun anvendes under graviditet, hvis det er klart nødvendigt, og hvis den potentielle fordel berettiger den potentielle risiko for fosteret.
Fodringstid
Det vides ikke, om dimethylfumarat eller dets metabolitter udskilles i modermælk. En risiko for de nyfødte / spædbørn kan ikke udelukkes. Der skal tages stilling til, om man skal afbryde amningen eller stoppe behandlingen med Tecfidera. Fordelen ved amning for barnet og fordelen ved behandling for kvinden skal overvejes.
Fertilitet
Der er ingen data om virkningerne af Tecfidera på menneskelig fertilitet. Data fra prækliniske undersøgelser tyder ikke på, at dimethylfumarat er forbundet med en øget risiko for nedsat fertilitet (se pkt. 5.3).
04.7 Virkninger på evnen til at føre motorkøretøj og betjene maskiner
Der er ikke udført undersøgelser af evnen til at føre motorkøretøj eller betjene maskiner.
04.8 Bivirkninger
Resumé af sikkerhedsprofilen
De mest almindelige bivirkninger (forekomst ≥10%) for patienter behandlet med Tecfidera var rødme (rødme) og gastrointestinale hændelser (dvs. diarré, kvalme, mavesmerter, smerter i øvre del af maven).Skylning og mave -tarmhændelser har en tendens til at opstå tidligt i behandlingen (især i løbet af den første måned), og hos patienter, der er tilbøjelige til rødme og gastrointestinale hændelser, kan disse hændelser fortsat forekomme intermitterende under hele behandlingen med Tecfidera. forekomst> 1%) hos patienter behandlet med Tecfidera var rødme (3%) og gastrointestinale hændelser (4%).
I placebokontrollerede og ukontrollerede kliniske forsøg modtog i alt 2.468 patienter Tecfidera og fulgte i op til 4 år med en samlet eksponering svarende til 3.588 personår. Cirka 1.056 patienter modtog mere end 2 års behandling. Med Tecfidera. Erfaring med ukontrollerede kliniske forsøg er i overensstemmelse med erfaring med placebokontrollerede kliniske forsøg.
Tabel over bivirkninger
Tabellen nedenfor viser de bivirkninger, der blev rapporteret hyppigere hos patienter behandlet med Tecfidera end hos patienter behandlet med placebo. Disse data stammer fra 2 afgørende fase 3, dobbeltblinde, placebokontrollerede kliniske forsøg med i alt 1.529 patienter behandlet med Tecfidera i op til 24 måneder med en samlet eksponering på 2.371 personår (se pkt.5.1 De beskrevne frekvenser i nedenstående tabel er baseret på 769 patienter behandlet med Tecfidera 240 mg to gange dagligt og 771 patienter behandlet med placebo.
Bivirkninger præsenteres i henhold til den medDRA anbefalede terminologi i MedDRA systemorganklasse. Forekomsten af de bivirkninger, der er anført nedenfor, udtrykkes i henhold til følgende konvention:
- Meget almindelig (≥1 / 10)
- Almindelig (≥1 / 100,
- Ikke almindelig (≥1 / 1.000,
- Sjælden (≥1 / 10.000,
- Meget sjælden (
- Ikke kendt (hyppigheden kan ikke estimeres ud fra de tilgængelige data)
Beskrivelse af udvalgte bivirkninger
Rødme (rødme)
I placebokontrollerede kliniske forsøg, forekomsten af rødme (skylning) (34% vs 4%) og hedeture (7% vs 2%) blev øget hos patienter behandlet med henholdsvis Tecfidera sammenlignet med dem, der blev behandlet med placebo. Skylning beskrives typisk som rødme eller hedeture, men kan omfatte andre hændelser (f.eks. Varme, rødme, kløe og en brændende fornemmelse). Skylningshændelser har en tendens til at forekomme tidligt i behandlingen (især i løbet af den første måned), og hos berørte patienter kan disse hændelser fortsat forekomme intermitterende under hele behandlingen med Tecfidera. af patienter, der blev behandlet med Tecfidera, afbrød behandlingen på grund af rødme.Forekomsten af alvorlig rødme, som kan være karakteriseret ved generaliseret erytem, udslæt og / eller kløe, blev observeret hos mindre end 1% af patienterne, der blev behandlet med Tecfidera (se pkt.4.2, 4.4 og 4.5).
Mave -tarmkanalen
Forekomsten af gastrointestinale hændelser (f.eks. Diarré [14% vs 10%], kvalme [12% vs 9%], smerter i øvre del af maven [10% vs 6%], mavesmerter [9% vs 4%], opkastning [8 % versus 5%] og dyspepsi [5% versus 3%]) blev forøget hos patienter behandlet med henholdsvis Tecfidera sammenlignet med dem, der blev behandlet med placebo. Gastrointestinale hændelser har tendens til at forekomme tidligt i behandlingen (især i løbet af den første måned) og hos ramte patienter, kan disse hændelser fortsat forekomme intermitterende under hele behandlingen med Tecfidera. Hos de fleste patienter, der oplevede gastrointestinale hændelser, var disse milde eller moderate i sværhedsgrad. 4% af patienterne, der blev behandlet med Tecfidera, afbrød behandlingen på grund af gastrointestinale hændelser. Forekomsten af alvorlige gastrointestinale hændelser, herunder gastroenteritis og gastritis, blev observeret hos 1% af patienterne behandlet med Tecfidera (se pkt.4.2).
Levertransaminaser
I placebokontrollerede undersøgelser blev stigninger i levertransaminaser observeret. Hos størstedelen af patienterne, hvor disse forhøjelser forekom, var levertransaminaser alaninaminotransferase og aspartataminotransferase (ASAT) ≥3 gange ULN, observeret hos henholdsvis 5% og 2% af placebo-behandlede patienter og hos 6% og 2% af patienter behandlet med Tecfidera Der blev ikke observeret nogen transaminaseforhøjelser ≥3 gange ULN med samtidige stigninger i total bilirubin> 2 gange ULN Afbrydelse af behandlingen på grund af forhøjede levertransaminaser var
Renal
I placebokontrollerede undersøgelser var forekomsten af proteinuri højere hos patienter behandlet med Tecfidera (9%) sammenlignet med placebo (7%) Den samlede forekomst af nyre- og urinbivirkninger var ens for patienter behandlet med Tecfidera og med placebo. Der er ikke rapporteret tilfælde af alvorlig nyreinsufficiens. Urinalyse viser, at procentdelen af patienter med proteinværdier på 1+ eller højere er ens for patienter behandlet med Tecfidera (43%) og patienter behandlet med placebo (40%). Typisk laboratorieobservationer af proteinuri De var ikke progressive. Sammenlignet til patienter behandlet med placebo blev der observeret en stigning i den estimerede glomerulære filtrationshastighed (eGFR) hos patienter behandlet med Tecfidera, inklusive dem, der oplevede 2 på hinanden følgende episoder med proteinuri (≥1 +).
Hæmatologisk
I placebokontrollerede kliniske forsøg var lymfocytværdier normale hos størstedelen af patienterne (> 98%) før behandlingsstart. Når behandlingen med Tecfidera blev påbegyndt, faldt gennemsnittet af lymfocyttal i løbet af det første år og stabiliserede sig derefter. I gennemsnit faldt lymfocyttallet cirka 30% fra baseline. Gennemsnitlige og mediane lymfocyttal forblev inden for normale grænser. Eosinofile lymfocyttal blev observeret i løbet af de første 2 måneder af behandlingen.
Laboratoriske abnormiteter
I placebokontrollerede kliniske forsøg var urinketonmålinger (1+ eller højere) overlegen hos patienter behandlet med Tecfidera (45%) sammenlignet med placebo (10%). Der blev ikke observeret uventede konsekvenser i kliniske undersøgelser.
1,25-dihydroxyvitamin D-niveauer faldt hos patienter behandlet med Tecfidera sammenlignet med dem, der blev behandlet med placebo (median procentvis fald fra baseline til 2 år med henholdsvis 25% sammenlignet med 15%) og parathyreoideahormon (PTH) blev øget hos patienter behandlet med Tecfidera sammenlignet med dem, der blev behandlet med placebo (stigning i medianprocenten fra baseline til 2 år på 29% sammenlignet med henholdsvis 15%). Middelværdier for begge parametre forblev inden for det normale område.
Indberetning af formodede bivirkninger
Rapportering af formodede bivirkninger, der opstår efter godkendelse af lægemidlet, er vigtig, da det muliggør løbende overvågning af lægemidlets nytte / risiko -forhold.Professionelle sundhedspersonale anmodes om at rapportere alle formodede bivirkninger via Det Italienske Lægemiddelagentur. . Websted: www.agenziafarmaco.gov.it/it/responsabili.
04.9 Overdosering
Der er ikke rapporteret tilfælde af overdosering.
05.0 FARMAKOLOGISKE EGENSKABER
05.1 Farmakodynamiske egenskaber
Farmakoterapeutisk gruppe: andre lægemidler i nervesystemet.
ATC -kode: N07XX09.
Handlingsmekanisme
Mekanismen, hvormed dimethylfumarat udøver terapeutiske virkninger ved multipel sklerose, er ikke fuldt ud forstået.Prækliniske undersøgelser viser, at de farmakodynamiske reaktioner af dimethylfumarat hovedsageligt medieres gennem aktivering af transkriptionsvejen til nuklear faktor Nrf2 (erythroid nuklear faktor 2 2-relateret). Det har vist sig, at dimethylfumarat forårsager opregulering hos patienter (opregulering) af Nrf2-afhængige antioxidantgener (f.eks. NAD (P) H dehydrogenase, quinon 1; [NQO1]).
Farmakodynamiske virkninger
Virkninger på immunsystemet
I prækliniske og kliniske undersøgelser viste Tecfidera antiinflammatoriske og immunmodulerende egenskaber. Dimethylfumarat og monomethylfumarat, hovedmetabolitten af dimethylfumarat, reducerede signifikant aktiveringen af immunceller og den efterfølgende frigivelse af proinflammatoriske cytokiner som reaktion på inflammatoriske stimuli i prækliniske modeller.I kliniske undersøgelser hos psoriasispatienter påvirkede dimethylfumarat lymfocytfænotyper ved nedregulering (nedregulering) af proinflammatoriske cytokinprofiler (TH1, TH17) og favoriserede produktionen af antiinflammatoriske cytokiner (TH2). Dimethylfumarat demonstrerede terapeutisk aktivitet i flere inflammatoriske og neuroinflammatoriske læsionsmodeller. I fase 3 -undersøgelser faldt gennemsnittet af lymfocyttal i gennemsnit med ca. 30% fra baseline i løbet af det første år med en efterfølgende stabiliseringsfase.
Virkning på det kardiovaskulære system
I et QTc -korrigeret interval (QTc) -studie havde enkeltdoser af Tecfidera 240 mg eller 360 mg sammenlignet med placebo ingen effekt på QTc -intervallet.
Klinisk effekt og sikkerhed
To randomiserede, dobbeltblinde, placebokontrollerede, 2-årige undersøgelser blev udført [Studie 1 (DEFINE) med 1.234 forsøgspersoner og Studie 2 (CONFIRM) med 1.417 forsøgspersoner] hos personer med recidiverende remitterende multipel sklerose (MS -RR). Ingen forsøgspersoner med progressive former for multipel sklerose blev inkluderet i disse undersøgelser. Effektivitet (se nedenstående tabel) og sikkerhed blev påvist hos personer med udvidet funktionsnedsættelsesstatus (EDSS) -scores fra 0 til 5 inklusive, som havde haft mindst 1 tilbagefald i løbet af "året før randomisering eller inden for 6 uger efter randomisering, de havde magnetisk resonansbilleddannelse af hjernen (MRI), der viste mindst en gadoliniumforstærkende (Gd +) læsion. Undersøgelse 2 omfattede en enkeltblind komparatorarm (raterblindet, dvs. undersøgelseslægen / undersøgeren, der vurderede behandlingsresponset i undersøgelsen, var i en blind tilstand) af behandling med glatirameracetat (GA).
I studie 1 havde patienterne følgende median baseline -karakteristika: alder 39 år, sygdomsvarighed 7,0 år, EDSS -score 2,0. Desuden havde 16% af patienterne en EDSS -score> 3,5, 28% havde ≥2 tilbagefald i det foregående år, og 42% havde tidligere modtaget andre godkendte behandlinger for multipel sklerose. I MR -kohorten var 36% af patienterne inkluderet i undersøgelse havde gadoliniumforstærkende læsioner (Gd +) ved baseline (gennemsnitligt antal Gd -læsioner + 1,4).
I undersøgelse 2 havde patienterne følgende baseline -karakteristika: alder 37 år, sygdomsvarighed 6,0 år, EDSS -score 2,5. Desuden havde 17% af patienterne en EDSS -score> 3,5, 32% havde ≥2 tilbagefald i det foregående år, og 30% havde tidligere modtaget andre godkendte behandlinger for multipel sklerose. I MR -kohorten var 45% af patienterne inkluderet i undersøgelse havde gadoliniumforstærkende læsioner (Gd +) ved baseline (gennemsnitligt antal Gd + læsioner 2,4).
Sammenlignet med placebo havde forsøgspersoner behandlet med Tecfidera en klinisk relevant og statistisk signifikant reduktion i: andelen af forsøgspersoner med tilbagefald efter 2 år, primært endepunkt i undersøgelse 1; den 2-årige årlige tilbagefaldshastighed, primære endepunkt for undersøgelse 2.
Den årlige tilbagefaldshastighed for glatirameracetat og placebo var henholdsvis 0,286 og 0,401 i undersøgelse 2, hvilket svarer til en reduktion på 29% (p = 0,013), hvilket er i overensstemmelse med godkendt forskrivningsinformation.
a Alle kliniske endepunktsanalyser blev foretaget ved intention-to-treat (ITT);
b MR -analyse anvendte MR -kohorten
* P -værdi
Effekt hos patienter med høj sygdomsaktivitet:
En konsekvent effekt af behandlingen på tilbagefald blev observeret hos en undergruppe af patienter med høj sygdomsaktivitet, mens effekten til tiden til vedvarende progression af handicap efter 3 måneder ikke var klart fastslået. På grund af undersøgelsesdesignet blev den "høje sygdomsaktivitet defineret som følger:
- Patienter med 2 eller flere tilbagefald om et år og med en eller flere Gadolinium-forstærkende (Gd) læsioner på hjernens magnetiske resonansbilleddannelse (MRI) (n = 42 i DEFINE-undersøgelsen; n = 51 i CONFIRM-undersøgelsen) eller ,
- Patienter, der ikke har reageret på et fuldstændigt og tilstrækkeligt forløb (mindst et års behandling) af beta-interferon, der har haft mindst 1 tilbagefald i det foregående år på behandling og mindst 9 hyperintense T2-læsioner på magnetisk resonansbilleddannelse (MR) ) i kraniet eller mindst en Gadolinium-forstærkende læsion (Gd) eller patienter med en uændret eller større tilbagefaldshastighed i det foregående år sammenlignet med de foregående 2 år (n = 177 i DEFINE-undersøgelsen; n = 141 i CONFIRM undersøgelse).
Pædiatrisk population
Det Europæiske Lægemiddelagentur har udsat forpligtelsen til at indsende resultaterne af undersøgelser med Tecfidera i en eller flere undergrupper af den pædiatriske population ved multipel sklerose (se afsnit 4.2 for oplysninger om pædiatrisk brug).
05.2 "Farmakokinetiske egenskaber
Administreret oralt undergår Tecfidera (dimethylfumarat) hurtig presystemisk esterasemedieret hydrolyse og omdannes til monomethylfumarat, dets vigtigste metabolit, som også er aktiv. Dimethylfumarat er ikke kvantificerbart i plasma efter oral administration af Tecfidera. Derfor er alle farmakokinetiske analyser relateret til dimethylfumarat blev udført med plasmakoncentrationer af monomethylfumarat Farmakokinetiske data blev opnået hos personer med multipel sklerose og hos raske frivillige.
Absorption
Tmax for monomethylfumarat er mellem 2 og 2,5 timer. Da Tecfidera gastro-resistente hårde kapsler indeholder mikrotabletter, som er beskyttet af en enterisk belægning, begynder absorptionen først, når de forlader maven (typisk mindre end 1 time). Efter administration med mad på 240 mg to gange dagligt, begynder median peak (Cmax) var 1,72 mg / l, og den samlede eksponering (AUC, areal under kurven) var 8,02 h.mg / l hos personer med multipel sklerose. Samlet set er C
max og AUC steg cirka på en dosisproportionel måde over det undersøgte dosisinterval (120 mg til 360 mg). Hos personer med multipel sklerose blev to doser på 240 mg administreret med 4 timers mellemrum over en periode på 4 timer. administration tre gange om dagen. Dette resulterede i minimal akkumulering af eksponering, hvilket resulterede i en stigning på 12% i median Cmax sammenlignet med dosering to gange dagligt (1,72 mg / L to gange dagligt vs 1,93 mg / L tre gange dagligt). Uden sikkerhedsmæssige konsekvenser.
Fødevarer har ingen klinisk signifikant virkning på dimethylfumarat -eksponering, men Tecfidera bør tages sammen med mad på grund af forbedret tolerabilitet for rødme eller gastrointestinale bivirkninger (se pkt. 4.2).
Fordeling
Det tilsyneladende fordelingsvolumen efter oral administration af Tecfidera 240 mg varierer fra 60 L til 90 L. Binding af monomethylfumarat til humane plasmaproteiner er generelt mellem 27% og 40%.
Biotransformation
Hos mennesker metaboliseres dimethylfumarat i vid udstrækning med mindre end 0,1% af den dosis, der udskilles i urinen som umodificeret dimethylfumarat.Dimethylfumarat metaboliseres i første omgang af esteraser, som er allestedsnærværende i mave -tarmkanalen, blod og væv, før de når den systemiske cirkulation. Yderligere metabolisme sker gennem tricarboxylsyre -cyklussen uden involvering af cytochrom P450 -systemet (CYP). En enkeltdosisundersøgelse af 240 mg 14C-dimethylfumarat identificerede glucose som den dominerende metabolit i humant plasma. Andre cirkulerende metabolitter omfattede fumarsyre, citronsyre og monomethylfumarat. Metabolismen af fumarsyre nedstrøms for den førnævnte metaboliske vej foregår gennem tricarboxylsyre -cyklussen med udånding af kuldioxid (CO2), der fungerer som den vigtigste eliminationsvej.
Eliminering
Udånding af CO2 er den vigtigste eliminationsvej for dimethylfumarat og tegner sig for 60% af dosis. Renal og fækal eliminering er sekundære eliminationsveje, der tegner sig for henholdsvis 15,5% og 0,9% af dosis.
Den terminale halveringstid for monomethylfumarat er kort (ca. 1 time), og der er ingen cirkulerende monomethylfumarat til stede ved 24 timer hos de fleste forsøgspersoner. Akkumulering af modermedicin eller monomethylfumarat forekommer ikke ved flere doser dimethylfumarat på det terapeutiske regime.
Linearitet
Eksponering for dimethylfumarat stiger på en cirka dosisproportional måde med enkelt- og multiple doser over det undersøgte dosisinterval på 120 mg til 360 mg.
Farmakokinetik i særlige patientgrupper
Baseret på resultaterne af variansanalysen (ANOVA) er kropsvægt den vigtigste eksponeringskovariat (ifølge Cmax og AUC) hos personer med tilbagefaldsgivende multipel sklerose (RRMS), men påvirkede ikke målingerne. Sikkerhed og effekt evalueret i kliniske forsøg.
Køn og alder havde ikke en klinisk signifikant indvirkning på dimethylfumaratets farmakokinetik.Farmakokinetik hos patienter 65 år og ældre er ikke undersøgt.
Pædiatrisk population
Farmakokinetik hos patienter under 18 år er ikke undersøgt.
Nedsat nyrefunktion
Da nyrevejen er en sekundær eliminationsvej for dimethylfumarat, der repræsenterer mindre end 16% af den administrerede dosis, er der ikke foretaget nogen evaluering af farmakokinetikken hos personer med nedsat nyrefunktion.
Nedsat leverfunktion
Da dimethylfumarat og monomethylfumarat metaboliseres af esteraser uden involvering af CYP450 -systemet, er der ikke blevet foretaget nogen evaluering af farmakokinetikken hos personer med nedsat leverfunktion.
05.3 Prækliniske sikkerhedsdata
Bivirkningerne beskrevet i afsnittene Toksikologi og reproduktionstoksicitet nedenfor blev ikke observeret i kliniske undersøgelser, men blev observeret hos dyr ved eksponeringsniveauer svarende til kliniske eksponeringsniveauer.
Mutagenese
Dimethylfumarat og mono-methylfumarat var negative i et enkelt testbatteri in vitro (Ames -test, test for kromosomafvigelser i pattedyrsceller). Dimethylfumarat var negativ i mikronukleustesten hos rotter in vivo.
Kræftfremkaldende
Carcinogenicitetsundersøgelser af dimethylfumarat blev udført i op til 2 år hos mus og rotter. Dimethylfumarat blev administreret oralt i doser på 25, 75, 200 og 400 mg / kg / dag til mus og i doser på 25, 50, 100 og 150 mg / kg / dag til rotter. Hos mus blev forekomsten af renalt tubulært carcinom øget ved en dosis på 75 mg / kg / dag, en tilsvarende eksponering (AUC) ved den anbefalede humane dosis. Hos rotter blev forekomsten af renal tubulært karcinom øget ved en dosis på 100 mg / kg / dag, en eksponering cirka 3 gange højere end den anbefalede humane dosis. Relevansen af disse fund for menneskelig risiko er ukendt.
Forekomsten af papillom og pladecellecarcinom i den ikke-glandulære del af maven (forestomach) blev øget ved en eksponering svarende til den anbefalede humane dosis hos mus og ved en eksponering under den anbefalede humane dosis til rotter (baseret på "AUC ). Der er ingen menneskelig pendant til gnavernes skovmage.
Toksikologi
Prækliniske undersøgelser blev udført på gnavere, kaniner og aber med en suspension af dimethylfumarat (dimethylfumarat i 0,8% hydroxypropylmethylcellulose) administreret ved oral sonde. Det kroniske hundeundersøgelse blev udført med oral administration af dimethylfumaratkapslen.
Nyreændringer blev observeret efter gentagen oral administration af dimethylfumarat i mus, rotter, hunde og aber. Der blev observeret regenerering af renalt tubulært epitel, der var tegn på skade, hos alle arter.Renal tubulær hyperplasi blev observeret hos rotter, der modtog livslang behandling (2-årigt studie). Kortikal atrofi blev observeret hos hunde og aber, og enkeltcellet nekrose og interstitiel fibrose blev observeret hos aber, der fik daglige orale doser dimethylfumarat i 12 måneder, 6 gange den anbefalede dosis baseret på AUC. Kender relevansen af disse fund for mennesker risiko.
Degeneration af det seminiferepitel blev observeret i testikler af rotter og hunde. Resultater blev observeret ved omtrent den anbefalede dosis til rotter og ved 6 gange den anbefalede dosis til hunde (baseret på AUC). Relevansen af disse fund for menneskelig risiko er ukendt.
Fundene i forestomach af mus og rotter var pladeepitelhyperplasi kombineret med hyperkeratose; betændelse; og papillom og pladecellecarcinom i undersøgelser, der varer 3 måneder eller længere. Der er ingen menneskelig pendant til skovsmagen af mus og rotter.
Reproduktionstoksicitet
Oral administration af dimethylfumarat til hanrotter på 75, 250 og 375 mg / kg / dag før og under parring havde ingen effekt på hanfrugtbarhed op til den højeste testede dosis (mindst 2x den anbefalede AUC -dosis.). Oral administration af dimethylfumarat til hunrotter med 25, 100 og 250 mg / kg / dag før og under parring og fortsatte indtil dag 7 i drægtigheden resulterede i en reduktion i antallet af østruscyklusser i 14 dage. Og øgede antallet af dyr på forlænget diestrus ved den højeste testede dosis (11 gange den anbefalede dosis baseret på AUC). Disse ændringer havde imidlertid ingen effekt på fertiliteten eller antallet af levedygtige fostre, der blev produceret.
Dimethylfumarat har vist sig at krydse placentamembranen og trænge ind i fosterblodet hos rotter og kaniner, med fostrets plasmakoncentration mellem foster og mor fra henholdsvis 0,48 til 0,64 og 0,1. Der blev ikke observeret misdannelser hos rotter eller kaniner ved nogen dosis dimethylfumarat. Administration af dimethylfumarat i orale doser på 25, 100 og 250 mg / kg / dag til drægtige rotter i organogeneseperioden gav moderens bivirkninger 4 gange den anbefalede dosis baseret på AUC og lav fostervægt og forsinket "ossifikation (metatarsal og bagbenets falanger) ved 11 gange den anbefalede dosis baseret på AUC. Lavere fostervægt og forsinket ossifikation blev betragtet som sekundær til moderens toksicitet (nedsat kropsvægt og fødeforbrug).
Oral administration af dimethylfumarat ved 25, 75 og 150 mg / kg / dag til gravide kaniner under organogenese havde ingen effekt på embryofoetal udvikling og resulterede i moderens vægt reduceret til 7 gange den anbefalede dosis og øget abort. Til 16 gange den anbefalede dosis, baseret på AUC.
Oral administration af dimethylfumarat ved 25, 100 og 250 mg / kg / dag til rotter under drægtighed og amning resulterede i reducerede kropsvægte i F1 kuld og forsinkelser i kønsmodning hos F1 hanner ved 11 gange den anbefalede dosis baseret på "AUC. Der var ingen effekt på fertiliteten i F1 -kuldene. Lavere kropsvægt af kuld blev betragtet som sekundær for moderens toksicitet.
06.0 LÆGEMIDDELOPLYSNINGER
06.1 Hjælpestoffer
Mikro-tabletter med enterisk belægning
Mikrokrystallinsk cellulose
Croscarmellosenatrium
Talc
Vandfri kolloid silica
Magnesiumstearat
Triethylcitrat
Methacrylsyre - methylmethacrylatcopolymer (1: 1)
Methacrylsyre - ethylacrylatcopolymer (1: 1) dispersion 30%
Simethicone
Natriumlaurylsulfat
Polysorbat 80
Kapselskal
Gele
Titandioxid (E171)
Strålende blå FCF (E133)
Gul jernoxid (E172)
Kapselprint (sort blæk)
Shellac
Kaliumhydroxid
Sort jernoxid (E172)
06.2 Uforenelighed
Ikke relevant.
06.3 Gyldighedsperiode
120 mg gastro-resistente hårde kapsler: 4 år.
240 mg gastro-resistente hårde kapsler: 3 år.
06.4 Særlige opbevaringsforhold
Må ikke opbevares over 30 ° C.
Opbevar vablerne i den ydre karton for at beskytte medicinen mod lys.
06.5 Den umiddelbare emballages art og emballagens indhold
120 mg kapsler: 14 kapsler i PVC / PE / PVDC-PVC aluminium blisterpakninger.
240 mg kapsler: 56 eller 168 kapsler i PVC / PE / PVDC-PVC aluminium blisterpakninger.
Ikke alle pakningsstørrelser er nødvendigvis markedsført.
06.6 Brugsanvisning og håndtering
Ingen særlige instruktioner.
07.0 INDEHAVER AF MARKEDSFØRINGSTILLADELSE
Biogen Idec Ltd.
Innovationshus
70 Norden Road
Maidenhead
Berkshire
SL6 4AY
Storbritannien
08.0 MARKEDSFØRINGSTILLADELSESNUMMER
A.I.C. n. 043217013 / E
A.I.C. n. 043217025 / E
09.0 DATO FOR FØRSTE TILLADELSE ELLER FORNYELSE AF TILLADELSEN
Dato for første godkendelse: 30. januar 2014
10.0 DATO FOR REVISION AF TEKSTEN
12/2015