Hvad er phenylketonuri
Der phenylketonuri (P.K.U.) det er en autosomal recessiv nedarvet metabolisk sygdom, der rammer 1 ud af 10.000 individer og ser ud til at forekomme mere i homozygositet end i heterozygoter.
Tilhørende til gruppen af hyperphenylalaninæmi kompromitterer phenylketonuri betydeligt metabolismen af phenylalanin og især dets konvertering til tyrosin; phenylketonuri genkendes af de forhøjede urinindhold af phenylalanin og nogle derivater (phenylpyruvat, phenylacetat, phenylactat og phenylacetylglutamin).
Den mest alvorlige komplikation af phenylketonuri er mental forsinkelse.
Phenylalanin, tyrosin og derivater
Phenylalanin er en essentiel aminosyre og udgør størstedelen af kosten proteiner; det kan omdannes af enzymet phenylalaninhydroxylase i tyrosin (ved tilsætning af en hydroxylgruppe -OH). Til gengæld er tyrosin en forløber -aminosyre til syntesen af:
- L-DOPA (dopaminsyntesemellemprodukt)
- Epinefrin
- Noradrenalin (alle neurotransmittere).

Mekanisme af phenylketonuri (P.K.U.)
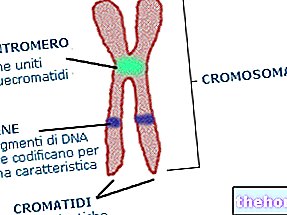
Som forventet er phenylketonuri på grund af en eller flere (6 i alt) kromosommutationer ekspressionen (derfor den metaboliske aktivitet) af phenylalaninhydroxylase praktisk talt nul. Disse ændringer kan være af forskellig art (fra "missense" -ændringer til "splejsning" -defekter eller endda "delvise sletninger"), men det er vigtigt, at på grund af denne enzymatiske ineffektivitet er phenylalaninniveauerne i blodet (som normalt er 1 mg / 100 ml) i DOMINANT phenylketonuri de når let mængder endda 50 gange højere.
Funktion af phenylalaninhydroxylaseenzymet: For at producere tyrosin (+ dihydrobiopterin) kræver phenylalaninhydroxylase: phenylalanin, oxygen og tetrahydrobiopterin (en reduceret pteridin, der fungerer som en coofactor); reaktionen er også reversibel, og dihydrobiopterin kan rekonverteres (takket være enzymet dihydropterin reduktase) i tetrahydrobiopterin.
Komplikationer
Fenylketonuri kan give anledning til mere eller mindre alvorlige komplikationer baseret på sværhedsgraden af den patologiske manifestation og rettidigheden af diagnosen; er en arvelig patologi, phenylketonuri skelnes i:
- Dominerende, derfor karakteriseret ved HELT inaktivitet af phenylalaninhydroxylaseenzymet
- Recessiv, hvor kun 30% af det samlede enzymatiske arv er aktivt.
Komplikationerne ved phenylketonuri skyldes og er direkte proportionale med den metaboliske ophobning af phenylalanin, dets derivater og den reducerede syntese af tyrosin.I patologi filtreres overskydende phenylalanin relativt effektivt af nyren, som kun delvist reabsorberer det, eliminerer det med urin ; persistensen af niveauerne af hyper-phenylalaninæmi bestemmer imidlertid en metabolisk reaktion af molekylær konvertering i phenylpyruvinsyre og / eller andre derivater lettere at dræne (phenylpyruvat, phenylacetat, phenylactat).
Det, der komplicerer phenylketonuri, er toksiciteten af phenylalanin, phenylpyruvinsyre og dets derivater over for centralnervesystemet (CNS). Deres overdrevne tilstedeværelse i hjernens udvikling bestemmer ubønhørligt en form for mental retardering.
NB. Plasmakoncentrationerne af de andre aminosyrer er lidt reduceret, sandsynligvis på grund af feedback på tarmabsorption eller renal tubulær reabsorption.
Hjerneskade, som en alvorlig komplikation af phenylketonuri, skyldes subtraktion af andre essentielle aminosyrer i proteosyntese, især i dannelsen af polyribosomer, myelin, noradrenalin og serotonin. Fenylketonuri - ikke synlig umiddelbart efter fødslen, men efter et par år - hvis det ikke behandles, kræver indlæggelse af barnet og er fuldstændig irreversibel.
Avanceret phenylketonuri kan også være tydeligt synligt for det blotte øje; de høje koncentrationer af phenylalanin, der hæmmer enzymet tyrosinase, forringer signifikant syntesen af melanin ved at reducere hud- og hårpigmentering; endvidere giver ophobningen af phenylacetat i håret og huden phenylketonurika en stærk og ubehagelig "muselugt".