Aktive ingredienser: Levetiracetam
Keppra 250 mg filmovertrukne tabletter
Keppra 500 mg filmovertrukne tabletter
Keppra 750 mg filmovertrukne tabletter
Keppra 1000 mg filmovertrukne tabletter
Keppra indlægssedler er tilgængelige til pakningsstørrelser: - Keppra 250 mg filmovertrukne tabletter, Keppra 500 mg filmovertrukne tabletter, Keppra 750 mg filmovertrukne tabletter, Keppra 1000 mg filmovertrukne tabletter
- Keppra 100 mg / ml oral opløsning
- Keppra 100 mg / ml koncentrat til infusionsvæske, opløsning
Hvorfor bruges Keppra? Hvad er det for?
Keppra er en antiepileptisk medicin (en medicin, der bruges til at behandle anfald).
Keppra bruges:
- alene hos voksne og unge fra 16 år med nydiagnosticeret epilepsi, til behandling af partielle anfald med eller uden sekundær generalisering.
- som et supplement til andre antiepileptika til behandling af:
- partielle anfald, med eller uden generalisering, hos voksne, unge, børn og spædbørn fra 1 måneders alder
- myokloniske anfald hos voksne og unge fra 12 år med ung myoklonisk epilepsi
- primære generaliserede tonisk-kloniske anfald hos voksne og unge fra 12 år med idiopatisk generaliseret epilepsi.
Kontraindikationer Når Keppra ikke bør bruges
Tag ikke Keppra
- Hvis du er allergisk (overfølsom) over for levetiracetam eller et af de øvrige indholdsstoffer i dette lægemiddel
Forholdsregler ved brug Hvad du skal vide, før du tager Keppra
Tal med din læge, før du tager Keppra
- Hvis du har nyreproblemer, skal du følge din læges instruktioner. Sidstnævnte kan beslutte, om dosis skal korrigeres.
- Hvis du bemærker en afmatning i væksten eller en uventet pubertetsudvikling hos dit barn, skal du kontakte din læge.
- Hvis du bemærker en stigning i anfaldets sværhedsgrad (f.eks. Øget antal), skal du kontakte din læge.
- Et begrænset antal mennesker, der behandles med antiepileptika som Keppra, har haft tanker om at skade eller tænke på selvmord. Hvis du har symptomer på depression og / eller selvmordstanker, skal du kontakte din læge.
Interaktioner Hvilke lægemidler eller fødevarer kan ændre virkningen af Keppra
Brug af anden medicin sammen med Keppra
Fortæl det til din læge eller apotek, hvis du tager eller for nylig har taget anden medicin, herunder medicin, der er købt uden recept.
Keppra sammen med mad, drikke og alkohol
Du kan tage Keppra med eller uden mad. Som en sikkerhedsforanstaltning må du ikke tage Keppra sammen med alkohol.
Advarsler Det er vigtigt at vide, at:
Graviditet og amning
Spørg din læge eller apotek til råds, før du tager medicin.
Fortæl det til din læge, hvis du er gravid eller tror, at du er gravid. Keppra bør ikke bruges under graviditet, medmindre det er klart nødvendigt. En risiko for fosterskader for fosteret kan ikke helt udelukkes.Keppra har vist uønskede reproduktionseffekter i dyreforsøg med højere dosisniveauer end dem, der er nødvendige for at kontrollere anfald.
Amning anbefales ikke under behandlingen.
Kørsel og brug af maskiner
Keppra kan reducere evnen til at køre bil eller bruge værktøjer eller maskiner, da Keppra kan gøre dig søvnig. Dette er mere sandsynligt i starten af behandlingen eller efter en dosisforøgelse. Du må ikke køre bil eller betjene maskiner, før du har kontrolleret, at din evne til at udføre disse aktiviteter ikke påvirkes.
Keppra 750 mg filmovertrukne tabletter indeholder Sunset Yellow FCF (E110)
Sunset Yellow FCF (E110) farvestof kan forårsage allergiske reaktioner. Andre styrker ved Keppra tabletter indeholder ikke denne komponent.
Dosis, metode og administrationstidspunkt Sådan bruges Keppra: Dosering
Tag altid dette lægemiddel nøjagtigt efter lægens eller apotekspersonalets anvisning.
Hvis du er i tvivl, skal du kontakte din læge eller apotek. Keppra skal tages to gange om dagen, en gang om morgenen og en gang om aftenen, på omtrent samme tid hver dag.
Tag antallet af tabletter i henhold til din læge instruktioner.
Monoterapi
- Dosis til voksne og unge (fra 16 år):
Typisk dosis: mellem 1000 mg og 3000 mg pr. Dag.
Når du begynder at tage Keppra for første gang, vil din læge ordinere en lavere dosis i 2 uger, før du giver dig den typiske lavere dosis.
Eksempel: hvis din daglige dosis er 1000 mg, kan du tage 2 tabletter à 250 mg om morgenen og 2 tabletter på 250 mg om aftenen.
Supplerende terapi
- Dosis til voksne og unge (12 til 17 år), der vejer 50 kg eller mere:
Typisk dosis: mellem 1000 mg og 3000 mg pr. Dag.
Eksempel: Hvis din daglige dosis er 1000 mg, kan du tage 2 tabletter à 250 mg om morgenen og 2 tabletter på 250 mg om aftenen.
- Dosis til spædbørn (6 til 23 måneder), børn (2 til 11 år) og unge (12 til 17 år), der vejer mindre end 50 kg:
Din læge vil ordinere den mest passende farmaceutiske form for Keppra afhængigt af din alder, vægt og dosis.
Keppra 100 mg / ml oral opløsning er den mest egnede præsentation til spædbørn og børn under 6 år.
Typisk dosis: mellem 20 mg pr. Kg legemsvægt og 60 mg pr. Kg legemsvægt pr. Dag.
Eksempel: For en typisk daglig dosis på 20 mg pr. Kg legemsvægt, hvis dit barn vejer 25 kg, kan du give ham 1 250 mg tablet om morgenen og 1 250 mg tablet om aftenen
- Dosis til spædbørn (1 måned til mindre end 6 måneder):
Keppra 100 mg / ml oral opløsning er en mere egnet præsentation til spædbørn.
Administration:
Synk Keppra tabletterne med en tilstrækkelig mængde væske (f.eks. Et glas vand).
Behandlingens varighed:
- Keppra bruges som en kronisk behandling. Keppra -behandling bør vare så længe din læge har bedt dig om det.
- Stop ikke behandlingen uden din læges råd, da dette kan øge antallet af anfald. Hvis din læge beslutter at stoppe Keppra -behandlingen, vil han instruere dig i gradvist at stoppe Keppra.
Overdosering Hvad skal jeg gøre, hvis du har taget for meget Keppra
Hvis du har taget for mange Keppra:
Mulige bivirkninger ved en overdosis Keppra er søvnighed, uro, aggression, nedsat opmærksomhed, vejrtrækningshæmning og koma. Kontakt din læge, hvis du har taget flere tabletter, end du burde. Din læge vil bestemme den bedst mulige behandling af overdosis.
Hvis du har glemt at tage Keppra:
Kontakt din læge, hvis du har glemt at tage en eller flere doser. Tag ikke en dobbeltdosis som erstatning for en glemt tablet
Hvis du holder op med at tage Keppra:
I tilfælde af afbrydelse af behandlingen, som med enhver anden antiepileptisk medicin, bør Keppra seponeres gradvist for at undgå en stigning i anfald.
Spørg din læge eller apotek, hvis du har yderligere spørgsmål om brugen af dette lægemiddel.
Bivirkninger Hvad er bivirkningerne af Keppra
Ligesom al anden medicin kan denne medicin forårsage bivirkninger, men ikke alle får bivirkninger. Nogle af bivirkningerne som søvnighed, træthed og svimmelhed kan være mere almindelige i starten af behandlingen eller når dosis øges, men disse effekter bør dog aftage med tiden.
Meget almindelig: kan forekomme hos mere end 1 ud af 10 patienter
- nasopharyngitis;
- døsighed, hovedpine.
Almindelig: kan forekomme hos 1 til 10 ud af 100 patienter
- anoreksi (tab af appetit);
- depression, fjendtlighed eller aggression, angst, søvnløshed, nervøsitet eller irritabilitet;
- kramper, balanceforstyrrelse, svimmelhed (ustabilitet), sløvhed, tremor (ufrivillige rystelser);
- svimmelhed (fornemmelse af rotation);
- hoste;
- mavesmerter, diarré, dyspepsi (fordøjelsesbesvær), opkastning, kvalme;
- udslæt;
- asteni / træthed (svaghed).
Ikke almindelig: kan forekomme hos 1 til 10 ud af 1000 patienter
- fald i antallet af blodplader i blodet, fald i antallet af hvide blodlegemer;
- vægttab, vægtforøgelse;
- selvmordsforsøg og selvmordstanker, psykisk lidelse, unormal adfærd, hallucinationer, vrede, forvirring, panikanfald, følelsesmæssig labilitet / humørsvingninger, uro;
- hukommelsestab (hukommelsestab), hukommelsesforringelse (glemsomhed), unormal koordination / ataksi (nedsat motorisk koordination), paræstesi (prikken), nedsat opmærksomhed (tab af koncentration);
- diplopi (dobbeltsyn), sløret syn;
- leverfunktionstest unormal;
- hårtab, eksem, kløe;
- muskelsvaghed, myalgi (muskelsmerter);
- trauma.
Sjælden: kan ramme 1 til 10 brugere ud af 10.000
- infektion;
- fald i antallet af alle typer blodlegemer
- alvorlige overfølsomhedsreaktioner (DRESS)
- fald i koncentrationen af natrium i blodet
- selvmord, personlighedsforstyrrelse (adfærdsproblemer), ændret tænkning (langsom tænkning, manglende koncentrationsevne);
- ukontrollerbare muskelspasmer, der involverer hoved, bagagerum og lemmer, vanskeligheder med at kontrollere bevægelser, hyperkinesis (hyperaktivitet);
- pancreatitis;
- leversvigt, hepatitis;
- hududslæt, der kan blære og fremstå som små mål (central mørk plet omgivet af et "lysere område, med en mørk ring rundt om kanten) (erythema multiforme), et udbredt udslæt med blærer og afskalning af huden, især omkring munden, næse, øjne og kønsorganer (Stevens-Johnsons syndrom) og en mere alvorlig form, der forårsager hudskalning i mere end 30% af kroppens overflade (toksisk epidermal nekrolyse).
Indberetning af bivirkninger
Tal med din læge eller apotek, hvis du får bivirkninger, herunder mulige bivirkninger, som ikke er nævnt i denne indlægsseddel. Du kan også rapportere bivirkninger direkte via det nationale rapporteringssystem. Ved at rapportere bivirkninger kan du hjælpe med at give mere information om sikkerheden ved dette lægemiddel.
Udløb og opbevaring
Opbevar denne medicin utilgængeligt for børn.
Brug ikke dette lægemiddel efter den udløbsdato, der står på kartonen efter EXP: og på blisterpakningen efter EXP:. Udløbsdatoen refererer til den sidste dag i den pågældende måned.
Denne medicin kræver ingen særlige opbevaringsbetingelser.
Smid ikke medicin via spildevand eller husholdningsaffald. Spørg din apotek om, hvordan du skal smide medicin, du ikke længere bruger. Dette vil hjælpe med at beskytte miljøet.
Pakningens indhold og andre oplysninger
Hvad Keppra indeholder
Den aktive ingrediens kaldes levetiracetam.
Hver Keppra 250 mg tablet indeholder 250 mg levetiracetam.
Hver Keppra 500 mg tablet indeholder 500 mg levetiracetam.
Hver Keppra 750 mg tablet indeholder 750 mg levetiracetam.
Hver Keppra 1000 mg tablet indeholder 1000 mg levetiracetam.
Øvrige indholdsstoffer er:
Tabletkerne: croscarmellosenatrium, macrogol 6000, vandfri kolloid silica, magnesiumstearat.
Belægning: delvist hydrolyseret polyvinylalkohol, titandioxid (E171), macrogol 3350, talkum, farvestoffer *.
* Farvestofferne er:
250 mg tabletter: indigo karmin aluminiumsø (E132)
500 mg tabletter: gult jernoxid (E172)
750 mg tabletter: solnedgangsgult FCF (E110), rødt jernoxid (E172)
1000 mg tabletter: (ingen ekstra farvestoffer).
Beskrivelse af hvordan Keppra ser ud og pakningens indhold
Keppra 250 mg filmovertrukne tabletter er blå, ovale formede, markerede og præget med "ucb" og "250" på den ene side.
Keppra 500 mg filmovertrukne tabletter er gule, ovale, skårne og præget med "ucb" og "500" på den ene side.
Keppra 750 mg filmovertrukne tabletter er orange, ovale formede, mærket og præget med "ucb" og "750" på den ene side.
Keppra 1000 mg filmovertrukne tabletter er hvide, ovale formede, markerede og præget med "ucb" og "1000" på den ene side.
Keppra tabletter er pakket i blisterpakninger anbragt i papkasser indeholdende:
- 250 mg: 20, 30, 50, 60, 100 x 1, 100 filmovertrukne tabletter og multipakninger indeholdende 200 (2 pakninger med 100) filmovertrukne tabletter.
- 500 mg: 10, 20, 30, 50, 60, 100 x 1, 100, 120 filmovertrukne tabletter og multipakninger indeholdende 200 (2 pakninger med 100) filmovertrukne tabletter.
- 50 mg: 20, 30, 50, 60, 80, 100 x 1, 100 filmovertrukne tabletter og multipakninger indeholdende 200 (2 pakninger med 100) filmovertrukne tabletter.
- 1000 mg: 10, 20, 30, 50, 60, 100 x 1, 100 filmovertrukne tabletter og multipakninger indeholdende 200 (2 pakninger med 100) filmovertrukne tabletter.
Pakninger med 100 x 1 tablet fås i aluminium / PVC perforerede enhedsdosisblister.
Alle andre pakninger fås i standard aluminium / PVC blister.
Ikke alle pakningsstørrelser er nødvendigvis markedsført
Indlægsseddel: AIFA (Italian Medicines Agency). Indhold offentliggjort i januar 2016. De foreliggende oplysninger er muligvis ikke opdaterede.
For at få adgang til den mest opdaterede version er det tilrådeligt at få adgang til webstedet AIFA (Italian Medicines Agency). Ansvarsfraskrivelse og nyttige oplysninger.
01.0 LÆGEMIDLETS NAVN
KEPPRA 500 MG TABLETTER OVERLAGET MED FILM
02.0 KVALITATIV OG KVANTITATIV SAMMENSÆTNING
Hver filmovertrukket tablet indeholder 500 mg levetiracetam.
Den fulde liste over hjælpestoffer findes i afsnit 6.1.
03.0 LÆGEMIDDELFORM
Filmovertrukket tablet.
Gul, ovalformet, indgraveret og med ordene "ucb" og "500" præget på den ene side.
04.0 KLINISKE OPLYSNINGER
04.1 Terapeutiske indikationer
Keppra er indiceret som monoterapi til behandling af partielle anfald med eller uden sekundær generalisering hos voksne og unge fra 16 år med nydiagnosticeret epilepsi.
Keppra er indiceret som supplerende behandling
• ved behandling af partielle anfald med eller uden sekundær generalisering hos voksne, unge, børn og spædbørn fra 1 måneders alder med epilepsi
• ved behandling af myokloniske anfald hos voksne og unge fra 12 år med juvenil myoklonisk epilepsi
• ved behandling af primære generaliserede tonisk-kloniske anfald hos voksne og unge fra 12 år med idiopatisk generaliseret epilepsi.
04.2 Dosering og indgivelsesmåde
Dosering
Monoterapi til voksne og unge fra 16 år
Den anbefalede startdosis er 250 mg to gange dagligt, som skal øges til en initial terapeutisk dosis på 500 mg to gange dagligt efter to uger. Dosis kan øges yderligere med 250 mg to gange dagligt hver anden uge baseret på det kliniske svar. Den maksimale dosis er 1500 mg to gange dagligt.
Tillægsbehandling til voksne (≥ 18 år) og unge (12 til 17 år), der vejer 50 kg eller mere
Den indledende terapeutiske dosis er 500 mg to gange dagligt. Denne dosis kan startes på den første behandlingsdag.
Baseret på den kliniske respons og tolerabilitet kan den daglige dosis øges op til maksimalt 1500 mg to gange dagligt. Dosisjusteringer kan foretages i 500 mg stigninger eller fald to gange dagligt hver anden til fjerde uge.
Særlige populationer
Ældre (65 år og derover)
Dosisjustering anbefales til ældre patienter med nedsat nyrefunktion (se "Nedsat nyrefunktion" nedenfor).
Nyresvigt
Den daglige dosis bør individualiseres i henhold til nyrefunktionen.
For voksne patienter henvises til følgende tabel og justeres doseringen som angivet. For at bruge denne doseringstabel er det nødvendigt at estimere patientens kreatininclearance (CLcr) i ml / min. CLcr i ml / min kan beregnes ud fra bestemmelsen af serumkreatinin (mg / dl) ved hjælp af følgende formel for voksne og unge, der vejer 50 kg eller mere:
Derudover justeres CLcr for kropsoverfladeareal (BSA) som følger:
Dosisjustering til voksne og unge patienter, der vejer mere end 50 kg med nedsat nyrefunktion:
En ladningsdosis på 750 mg anbefales på den første behandlingsdag med levetiracetam.
Efter dialyse anbefales en ekstra dosis på mellem 250 og 500 mg.
For børn med nedsat nyrefunktion skal dosis af levetiracetam justeres baseret på nyrefunktionen, da levetiracetams clearance er relateret til nyrefunktionen. Denne anbefaling er baseret på en undersøgelse udført med voksne patienter med nedsat nyrefunktion.
Hos unge unge, børn og spædbørn kan CLcr i ml / min / 1,73 m2 estimeres ud fra bestemmelsen af serumkreatinin (i mg / dl) ved hjælp af følgende formel (Schwartz's formel):
ks = 0,45 hos spædbørn i alderen op til 1 år; ks = 0,55 hos børn under 13 år og hos unge kvinder; ks = 0,7 hos unge mænd.
Dosisjustering til spædbørn, børn og unge, der vejer mindre end 50 kg med nedsat nyrefunktion:
Keppra oral opløsning bør bruges til doser under 250 mg og til patienter, der ikke kan synke tabletter.
En 10,5 mg / kg (0,105 ml / kg) ladningsdosis anbefales på den første behandlingsdag med levetiracetam.
En 15 mg / kg (0,15 ml / kg) ladningsdosis anbefales på den første behandlingsdag med levetiracetam.
Efter dialyse anbefales en supplerende dosis på 3,5 til 7 mg / kg (0,035 til 0,07 ml / kg).
Efter dialyse anbefales en yderligere dosis på 5 til 10 mg / kg (0,05 til 0,10 ml / kg).
Leverinsufficiens
Ingen dosisjustering er nødvendig hos patienter med let til moderat nedsat leverfunktion. Hos patienter med alvorlig leverinsufficiens kan kreatininclearance undervurdere graden af nyreinsufficiens.Derfor anbefales en reduktion på 50% af den daglige vedligeholdelsesdosis, når kreatininclearance er 2.
Pædiatrisk population
Lægen bør ordinere den mest passende farmaceutiske form og styrke baseret på alder, vægt og dosis.
Tabletformuleringen er ikke egnet til brug hos spædbørn og børn under 6 år. Keppra oral opløsning er den foretrukne formulering til brug i denne population. Desuden er tabletternes tilgængelige styrker ikke egnede til indledende behandling hos børn, der vejer mindre end 25 kg, til patienter, der ikke kan synke tabletter eller til at administrere doser under 250 mg. I alle ovennævnte tilfælde bør Keppra oral opløsning anvendes.
Monoterapi
Sikkerheden og effekten af Keppra givet som monoterapi til børn og unge under 16 år er ikke klarlagt.
Ingen data er tilgængelige.
Tillægsbehandling til spædbørn i alderen 6 til 23 måneder, børn (2 til 11 år) og unge (12 til 17 år), der vejer mindre end 50 kg
Keppra oral opløsning er den foretrukne formulering til brug hos spædbørn og børn under 6 år.
Den indledende terapeutiske dosis er 10 mg / kg to gange dagligt.
Baseret på den kliniske respons og tolerabilitet kan dosis øges op til 30 mg / kg to gange dagligt. Dosisjusteringer bør ikke overstige stigninger eller fald på 10 mg / kg to gange dagligt hver anden uge. Den laveste effektive dosis bør anvendes.
Dosis til børn, der vejer 50 kg eller mere, er den samme som hos voksne.
Anbefalet dosis til spædbørn fra 6 måneder, børn og unge:
Børn, der vejer 25 kg eller mindre, skal helst starte med behandling med Keppra 100 mg / ml oral opløsning.
Dosis til børn og unge, der vejer 50 kg eller mere, er den samme som hos voksne.
Tillægsbehandling til spædbørn fra 1 måned til under 6 måneders alder
Den orale opløsning er formuleringen til brug hos spædbørn.
Indgivelsesmåde
De filmovertrukne tabletter skal administreres oralt, synkes med en tilstrækkelig mængde væske og kan tages med eller uden mad. Den daglige dosis skal deles i to i to administrationer.
04.3 Kontraindikationer
Overfølsomhed over for det aktive stof eller over for andre pyrrolidonderivater eller over for et eller flere af hjælpestofferne anført i pkt.6.1.
04.4 Særlige advarsler og passende forholdsregler ved brug
Afbrydelse af behandlingen
I overensstemmelse med den nuværende kliniske praksis anbefales en gradvis tilbagetrækning, hvis behandlingen med Keppra skal afbrydes (f.eks. Hos voksne og unge, der vejer mere end 50 kg: fald med 500 mg to gange dagligt med intervaller mellem to og fire uger; hos spædbørn over 6 måneders alder, hos børn og unge, der vejer mindre end 50 kg: dosisreduktion må ikke overstige 10 mg / kg to gange dagligt hver anden uge; hos spædbørn (under 6 måneder): dosisreduktion bør ikke overstige 7 mg / kg to gange om dagen hver anden uge).
Nyresvigt
Administration af Keppra til patienter med nedsat nyrefunktion kan kræve dosisjustering. Hos patienter med svært nedsat leverfunktion anbefales det at evaluere nyrefunktionen, inden doseringen etableres (se pkt.4.2).
Selvmord
Tilfælde af selvmord, selvmordsforsøg, selvmordstanker og adfærd er blevet rapporteret hos patienter behandlet med antiepileptika (herunder levetiracetam). En metaanalyse af randomiserede, placebokontrollerede forsøg med antiepileptiske lægemidler viste en let øget risiko for selvmordstanker og adfærd. Mekanismen for denne risiko kendes ikke.
Derfor bør patienter overvåges for tegn på depression og / eller selvmordstanker og adfærd, og passende behandling bør overvejes. Patienter (og pårørende) bør informeres om, at hvis der opstår tegn på depression og / eller selvmordstanker eller adfærd, skal læge søges.
Pædiatrisk population
Tabletformuleringen er ikke egnet til brug hos spædbørn og børn under 6 år.
Tilgængelige data om børn tyder ikke på en indflydelse på vækst og pubertet. Langtidseffekterne på læring, intelligens, vækst, hormonfunktion, pubertet og reproduktionspotentiale hos børn er imidlertid ukendte.
Sikkerhed og effekt af levetiracetam er ikke blevet grundigt evalueret hos spædbørn under 1 år med epilepsi.I kliniske undersøgelser blev kun 35 spædbørn i alderen <1 år med partielle anfald udsat for Keppra, hvoraf kun 13 var under en alder af 6 måneder.
04.5 Interaktioner med andre lægemidler og andre former for interaktion
Antiepileptika
Data fra præ-marketing kliniske undersøgelser foretaget hos voksne viser, at Keppra ikke påvirker serumkoncentrationerne af eksisterende antiepileptika (phenytoin, carbamazepin, valproinsyre, phenobarbital, lamotrigin, gabapentin og primidon), og at disse antiepileptika ikke påvirker farmakokinetikken for Keppra.
Som hos voksne er der hos pædiatriske patienter, der får levetiracetam -doser op til 60 mg / kg / dag, ingen tegn på klinisk signifikante interaktioner med andre lægemidler.
En retrospektiv evaluering af farmakokinetiske interaktioner hos børn og unge med epilepsi (4 til 17 år) bekræftede, at tillægsbehandling med oralt administreret levetiracetam ikke påvirkede steady-state serumkoncentrationer af carbamazepin og valproat administreret samtidigt. Data tyder imidlertid på en 20% højere levetiracetam -clearance hos børn, der tager enzymfremkaldende antiepileptiske lægemidler. Ingen dosisjustering er nødvendig.
Probenecid
Probenecid (500 mg fire gange dagligt), et renalt tubulært sekretionsblokerende middel, har vist sig at hæmme renal clearance af den primære metabolit, men ikke af levetiracetam. Imidlertid er koncentrationen af denne metabolit stadig lav. Andre lægemidler, der udskilles med aktiv tubulær sekretion, forventes at reducere metabolitens renale clearance. Levetiracetams virkning på probenecid er ikke undersøgt, og levetiracetams virkning på andre aktivt udskillede lægemidler, f.eks. NSAID'er, sulfonamider og methotrexat, er ukendt.
Orale præventionsmidler og andre farmakokinetiske interaktioner
Levetiracetam 1000 mg dagligt påvirkede ikke farmakokinetikken af orale præventionsmidler (ethinylestradiol og levonorgestrel); de endokrine parametre (luteiniserende hormon og progesteron) blev ikke ændret. Levetiracetam 2000 mg dagligt påvirkede ikke digoxins og warfarins farmakokinetik; protrombintiden blev ikke ændret. Samtidig administration af digoxin, orale præventionsmidler og warfarin påvirkede ikke levetiracetams farmakokinetik.
Antacida
Der er ingen data tilgængelige om antacida's indflydelse på absorptionen af levetiracetam.
Afføringsmidler
Der har været isolerede rapporter om nedsat effekt af levetiracetam, når det osmotiske afføringsmiddel macrogol blev administreret samtidigt med oralt levetiracetam. Derfor bør macrogol ikke tages oralt mellem en time før og en time efter indtagelse af levetiracetam.
Mad og alkohol
Omfanget af levetiracetamabsorption blev ikke påvirket af mad, men absorptionshastigheden blev reduceret en smule.
Der er ingen data om interaktionen mellem levetiracetam og alkohol.
04.6 Graviditet og amning
Graviditet
Data efter markedsføring fra flere potentielle graviditetsregistre har dokumenteret resultaterne af eksponering for levetiracetam som monoterapi hos mere end 1000 kvinder i graviditetens første trimester. Samlet set tyder disse data ikke på en væsentlig stigning i risikoen for større medfødte misdannelser, selvom en teratogen risiko ikke helt kan udelukkes. Terapi med flere AED'er er forbundet med en højere risiko for medfødte misdannelser end monoterapi, og derfor bør monoterapi overvejes. Dyrestudier har vist reproduktionstoksicitet (se pkt. 5.3).
Keppra anbefales ikke, medmindre det er klinisk nødvendigt, under graviditet og hos kvinder i den fertile alder, der ikke anvender præventionsmetoder.
Som med andre antiepileptika kan fysiologiske ændringer i forbindelse med graviditet påvirke plasmakoncentrationen af levetiracetam. Under graviditeten blev der observeret nedsatte plasmakoncentrationer af levetiracetam. Denne reduktion er mest markant i tredje trimester (op til 60% af baseline -koncentrationen før graviditet). Gravide kvinder, der behandles med levetiracetam, bør følges nøje fra et klinisk synspunkt. Afbrydelse af antiepileptiske behandlinger kan føre til en forværring af sygdommen, som kan være skadelig for moderen og fosteret.
Fodringstid
Levetiracetam udskilles i human modermælk. Derfor anbefales amning ikke.Hvis behandling med levetiracetam bliver nødvendig under amning, skal fordel / risiko -forholdet af behandlingen vejes under hensyntagen til amningens betydning.
Fertilitet
Der blev ikke fundet nogen indvirkning på fertiliteten i dyreforsøg (se pkt. 5.3). Der foreligger ingen kliniske data; den potentielle risiko hos mennesker er ukendt.
04.7 Virkninger på evnen til at føre motorkøretøj og betjene maskiner
Der er ikke udført undersøgelser af evnen til at føre motorkøretøj eller betjene maskiner.
I betragtning af den mulige forskellige individuelle følsomhed kan nogle patienter opleve søvnighed eller andre symptomer relateret til virkningen på centralnervesystemet, især i starten af behandlingen eller efter en stigning i dosis. Derfor anbefales forsigtighed hos patienter, der er involveret i aktiviteter, der kræver høj koncentration, såsom at køre køretøjer eller betjene maskiner. Patienter bør rådes til ikke at køre bil eller betjene maskiner, før det er fastslået, at deres evne til at udføre disse aktiviteter er upåvirket.
04.8 Bivirkninger
Resumé af sikkerhedsprofilen
Bivirkningsprofilen vist nedenfor er baseret på analysen af poolede placebokontrollerede kliniske forsøg på tværs af alle undersøgte indikationer for i alt 3416 patienter behandlet med levetiracetam. Disse data suppleres med brug af levetiracetam i tilsvarende åbne forlængelsesundersøgelser samt fra markedsføring. De hyppigst rapporterede bivirkninger var nasopharyngitis, søvnighed, hovedpine, træthed og svimmelhed.Sikkerhedsprofilen for levetiracetam er generelt ens på tværs af aldersgrupper (voksne og pædiatriske patienter) og de godkendte indikationer for behandling af epilepsi.
Tabel over bivirkninger
Bivirkninger rapporteret fra kliniske forsøg (voksne, unge, børn og spædbørn over 1 måned) og efter markedsføring er angivet i følgende tabel efter systemorganklasse og hyppighed. Er defineret som følger: meget almindelig (≥1 / 10); almindelig (≥1 / 100,
Beskrivelse af udvalgte bivirkninger
Risikoen for anoreksi er højere, når topiramat administreres samtidigt med levetiracetam.
I mange tilfælde af alopeci er helbredelse observeret efter afbrydelse af levetiracetam -behandlingen.
Knoglemarvsundertrykkelse blev identificeret i nogle af tilfældene med pancytopeni.
Pædiatrisk population
Hos patienter i alderen 1 måned til mindre end 4 år blev i alt 190 patienter behandlet med levetiracetam i placebokontrollerede og åbne forlængelsesundersøgelser. 60 af disse patienter blev behandlet med levetiracetam i placebokontrollerede undersøgelser. Hos patienter i alderen 4 til 16 år blev i alt 645 patienter behandlet med levetiracetam i placebokontrollerede og åbne forlængelsesstudier. 233 af disse patienter blev behandlet med levetiracetam i placebokontrollerede undersøgelser. I begge disse pædiatriske aldersgrupper er disse data integreret med erfaring efter markedsføring med brug af levetiracetam.
Bivirkningsprofilen for levetiracetam er generelt ens på tværs af aldersgrupper og på tværs af de godkendte epilepsiindikationer. I placebokontrollerede kliniske forsøg var sikkerhedsresultaterne hos pædiatriske patienter i overensstemmelse med sikkerhedsprofilen for levetiracetam hos voksne med undtagelse af adfærdsmæssige og psykiatriske bivirkninger, der var mere almindelige hos børn end hos voksne. Hos børn og unge i alderen 4-16 år blev opkastning (meget almindelig, 11,2%), uro (almindelig, 3,4%) rapporteret hyppigere end i andre aldersgrupper eller i den overordnede sikkerhedsprofil.), Humørsvingninger (almindelig, 2,1 %), affektiv labilitet (almindelig, 1,7%), aggression (almindelig, 8,2%), unormal adfærd (almindelig, 5,6%) og sløvhed (almindelig, 3,9%) Hos spædbørn og børn i alderen 1 måned til under 4 år, irritabilitet blev rapporteret hyppigere end i andre aldersgrupper eller i den overordnede sikkerhedsprofil (meget almindelig, 11,7%) og unormal koordination (almindelig, 3,3%).
En sikkerhedsundersøgelse hos pædiatriske patienter, udført i henhold til et ikke-mindreværdigt, dobbeltblindet, placebokontrolleret design, evaluerede de kognitive og neuro-psykologiske virkninger af Keppra hos børn i alderen 4 til 16 år med partielle anfald. Keppra viste sig ikke at være anderledes (ikke ringere) end placebo i ændringen fra baseline i scoren opnået i "Attention and Memory" subtest of Leiter-R scale (Memory Screen Composite score) i befolkningen pr. protokol. Resultaterne relateret til adfærdsmæssige og følelsesmæssige funktioner indikerede en forværring af aggressiv adfærd målt på en standardiseret og systematisk måde hos patienter behandlet med Keppra (brug af et valideret værktøj (CBCL - Achenbach børnes adfærdstjekliste). Emner, der tog Keppra i den åbne, langsigtede opfølgningsundersøgelse, oplevede imidlertid ikke i gennemsnit forringelse af deres adfærdsmæssige og følelsesmæssige funktioner; især blev vurderingerne af aggression i adfærd ikke forringet i forhold til baseline.
04.9 Overdosering
Symptomer
Somnolens, uro, aggression, nedsat bevidsthedsniveau, respirationsdepression og koma er observeret med Keppra -overdoser.
Behandling af overdosering
Efter en akut overdosis kan maven tømmes ved gastrisk skylning eller fremkaldelse af opkastning. Der er ingen specifik modgift mod levetiracetam. Behandling af overdosis levetiracetam bør være symptomatisk og kan omfatte hæmodialyse Ekstraktionseffektiviteten ved dialyse er 60% for levetiracetam og 74% for den primære metabolit.
05.0 FARMAKOLOGISKE EGENSKABER
05.1 Farmakodynamiske egenskaber
Farmakoterapeutisk gruppe: antiepileptika, andre antiepileptika, ATC -kode: N03AX14.
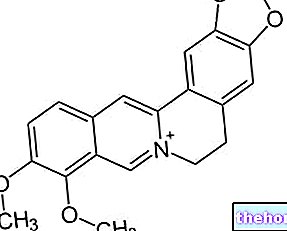
Det aktive stof, levetiracetam, er et pyrrolidonderivat (S-enantiomer af α-ethyl-2-oxo-1-pyrrolidinacetamid), kemisk uafhængigt af eksisterende antiepileptiske stoffer.
Handlingsmekanisme
Levetiracetams virkningsmekanisme er endnu ikke fuldstændigt forklaret, men det ser ud til at være forskellig fra mekanismerne for nuværende antiepileptika. in vitro og in vivo tyder på, at levetiracetam ikke ændrer grundlæggende cellulære egenskaber og normal neurotransmission.
Uddannelse in vitro viser, at levetiracetam virker på intraneuronale niveauer af Ca2 + ved delvis at hæmme N-type Ca2 + -strømme og ved at reducere frigivelsen af Ca2 + fra intraneuronale lagringssteder. Derudover vender det delvist reduktionen, induceret af zink og β-carbolin, af strømmen induceret af GABA og glycin. Uddannelse in vitro de fandt også ud af, at levetiracetam binder sig til et bestemt sted i gnaverens hjernevæv. Dette bindingssted er synaptisk vesikelprotein 2A, som menes at være involveret i vesikelfusion og neurotransmitter -exocytose. Levetiracetam og beslægtede analoger viser en grad af affinitet for binding til synaptisk vesikelprotein 2A, der korrelerer med styrken af deres antiepileptiske beskyttelse i det audiogene model for epilepsi hos mus. Dette fund tyder på, at interaktionen mellem levetiracetam og synaptisk vesikelprotein 2A ser ud til at spille en rolle i lægemidlets mekanisme for antiepileptisk virkning.
Farmakodynamiske virkninger
Levetiracetam fremkalder beskyttende virkning i et bredt spektrum af dyremodeller af delvis og primær generaliseret epilepsi uden at have en pro-krampagtig virkning.Den primære metabolit er inaktiv.
Hos mennesker bekræftede aktivitet i både partielle og generaliserede epilepsitilstande (epileptisk udladning / fotoparoksysmal respons) det brede spektrum af den farmakologiske profil af levetiracetam.
Klinisk effekt og sikkerhed
Supplerende behandling til behandling af partielle anfald med eller uden sekundær generalisering hos voksne, unge, børn og spædbørn fra 1 måneders alder med epilepsi.
Hos voksne blev levetiracetams effekt vist i 3 dobbeltblinde, placebokontrollerede undersøgelser med doser på 1000 mg, 2000 mg eller 3000 mg / dag, opdelt i 2 doser, i en behandlingsvarighed på op til 18 uger. analyse procentdelen af patienter, der opnåede en reduktion i frekvensen af partielle anfald pr. uge, i behandlingsperioden med stabil dosis (12/14 uger), svarende til eller større end 50% fra baseline, var 27, 7%, 31,6% og 41,3 % af patienterne behandlet med henholdsvis 1000, 2000 eller 3000 mg levetiracetam og 12,6% for patienter behandlet med placebo.
Pædiatrisk population
Levetiracetams effekt hos pædiatriske patienter (4 til 16 år) blev påvist i et dobbeltblindet, placebokontrolleret studie, der omfattede 198 patienter og havde en behandlingsvarighed på 14 uger. I denne undersøgelse modtog patienter levetiracetam ved en fast dosis på 60 mg / kg / dag (to gange dagligt).
44,6% af levetiracetam-behandlede patienter og 19,6% af placebo-behandlede patienter havde en reduktion på 50% eller større i partiel anfaldsfrekvens om ugen fra baseline. Ved fortsat langtidsbehandling var 11,4% af patienterne anfaldsfrie i mindst 6 måneder og 7,2% var anfaldsfrie i mindst 1 år.
Hos pædiatriske patienter (1 måned til mindre end 4 år) blev levetiracetams effekt påvist i et dobbeltblindet, placebokontrolleret studie, der omfattede 116 patienter og havde en behandlingsvarighed på 5 dage. blev ordineret en daglig dosis på 20 mg / kg, 25 mg / kg, 40 mg / kg eller 50 mg / kg oral opløsning baseret på deres aldersrelaterede dosistitreringsplan. følgende doser blev brugt: 20 mg / kg / dag , titreret til 40 mg / kg / dag, for spædbørn fra en måned til mindre end seks måneders alder; 25 mg / kg / dag, titreret til 50 mg / kg / dag for spædbørn og børn fra 6 måneder til mindre end 4 år af alder Den samlede daglige dosis blev opdelt i to administrationer pr. dag.
Hovedmålet for behandlingseffekt var antallet af patienter, der reagerede (procentdel af patienter med ≥50% reduktion i gennemsnitlig daglig frekvens af partielle anfald fra baseline), vurderet af en enkelt blindet undersøger, der brugte video -EEG i en periode på 48 timer. Effektivitetsanalysen blev udført på 109 patienter, der havde gennemgået video-EEG i mindst 24 timer, både i basisperioden og i evalueringsperioden. 43,6% af patienterne Levetiracetam-behandlede patienter og 19,6% af placebo-behandlede patienter blev anset for at være responsive. Resultaterne var konsistente på tværs af aldersgrupper. Ved fortsat langtidsbehandling var 8,6% af patienterne anfaldsfrie i mindst 6 måneder og 7,8% var anfaldsfrie i mindst 1 år.
Monoterapi til behandling af partielle anfald med eller uden sekundær generalisering hos patienter fra 16 år med nydiagnosticeret epilepsi.
Effekten af levetiracetam som monoterapi blev påvist i et dobbeltblindet, parallelt grupperet sammenlignende non-inferioritetsstudie versus kontrolleret frigivelse af carbamazepin (CR) hos 576 patienter i alderen 16 år eller ældre med ny eller ny epilepsi. For nylig diagnosticeret. har kun uprovokerede partielle anfald eller generaliserede tonisk -kloniske anfald Patienter blev randomiseret til carbamazepin CR 400 - 1200 mg / dag eller levetiracetam 1000 - 3000 mg / dag og behandlingen varede op til 121 uger baseret på respons.
Beslaglæggelsesfrihed i en periode på 6 måneder blev opnået hos 73,0% af patienterne behandlet med levetiracetam og hos 72,8% af patienterne behandlet med carbamazepin CR; den korrigerede absolutte forskel mellem behandlingerne var 0,2% (95% CI: 7,8 - 8,2). Mere end halvdelen af forsøgspersonerne forblev anfaldsfrie i 12 måneder (56,6% og 58,5% af forsøgspersonerne behandlet med henholdsvis levetiracetam og carbamazepin CR).
I en undersøgelse, der afspejler klinisk praksis, kan samtidig antiepileptisk behandling seponeres hos et begrænset antal patienter, der reagerede på levetiracetam-tillægsbehandling (36 ud af 69 voksne patienter).
Supplerende behandling til behandling af myokloniske anfald hos voksne og unge fra 12 år med juvenil myoklonisk epilepsi.
Levetiracetams effekt blev påvist i et 16-ugers, dobbeltblindet, placebokontrolleret studie med patienter i alderen 12 år eller ældre med idiopatisk generaliseret epilepsi med myokloniske anfald i forskellige syndromer. Størstedelen af patienterne havde juvenil myoklonisk epilepsi.
I denne undersøgelse var levetiracetam -dosis 3000 mg / dag givet i to opdelte doser.
58,3% af levetiracetam-behandlede patienter og 23,3% af placebo-behandlede patienter havde mindst 50% reduktion i myokloniske anfaldsdage om ugen. Efter fortsat langtidsbehandling var 28,6% af patienterne fri for myokloniske anfald i mindst 6 måneder, og 21,0% af patienterne var fri for myokloniske anfald i mindst 1 år.
Supplerende behandling til behandling af primære generaliserede tonisk-kloniske anfald hos voksne og unge fra 12 år med idiopatisk generaliseret epilepsi.
Levetiracetams effekt blev påvist i et 24-ugers dobbeltblindet, placebokontrolleret studie, der omfattede voksne, unge og et begrænset antal børn med idiopatisk generaliseret epilepsi med primære generaliserede tonisk-kloniske anfald (PGTC'er) i forskellige syndromer ( juvenil myoklonisk epilepsi, ungdomsfraværsepilepsi, infantil fraværsepilepsi eller epilepsi med anfald af stor mand ved opvågning) .I denne undersøgelse var dosis levetiracetam 3000 mg / dag for voksne og unge eller 60 mg / kg / dag for børn givet i to opdelte doser.
72,2% af levetiracetam-behandlede patienter og 45,2% af placebo-behandlede patienter havde en reduktion i PGTC-anfaldsfrekvensen på 50% eller mere om ugen. Efter fortsat langtidsbehandling var 47,4% af patienterne fri for tonisk-kloniske anfald i mindst 6 måneder og 31,5% var fri for tonisk-kloniske anfald i mindst 1 år.
05.2 Farmakokinetiske egenskaber
Levetiracetam er en meget opløselig og permeabel forbindelse. Den farmakokinetiske profil er lineær med ringe intra- og interindividuel variation. Der er ingen ændring i clearance efter gentagen administration. Der er ingen tegn på relevant døgnrytme og køn og race -variation. Den farmakokinetiske profil er sammenlignelig hos raske frivillige og hos patienter med epilepsi.
I betragtning af dets fuldstændige og lineære absorption kan plasmaniveauer af levetiracetam forudsiges ud fra den orale dosis udtrykt som mg / kg legemsvægt. Derfor er det ikke nødvendigt at overvåge levetiracetams plasmaniveauer.
Der var en signifikant sammenhæng mellem spyt- og plasmakoncentrationer hos voksne og børn (forholdet mellem spyt / plasmakoncentrationer varierede fra 1 til 1,7 for den orale tabletformulering og, efter 4 timer fra "indtagelse, til oral opløsning).
Voksne og unge
Absorption
Levetiracetam absorberes hurtigt efter oral administration. Oral biotilgængelighed er tæt på 100%.
Højeste plasmakoncentration (Cmax) nås 1,3 timer efter dosering. Steady-state opnås efter to dage med to daglige doser.
Højeste plasmakoncentrationer (Cmax) er typisk 31 og 43 mcg / ml efter henholdsvis en enkelt dosis på 1000 mg og gentaget 1000 mg to gange dagligt.
Absorptionsgraden er ikke dosisafhængig og påvirkes ikke af mad.
Fordeling
Der er ingen data om vævsfordeling hos mennesker.
Hverken levetiracetam eller dets primære metabolit binder sig signifikant til plasmaproteiner (
Distributionsvolumenet for levetiracetam er cirka 0,5 til 0,7 l / kg og er tæt på den samlede mængde vand.
Biotransformation
Levetiracetam metaboliseres ikke i udstrakt grad hos mennesker. Den vigtigste metaboliske vej (24% af dosis) er enzymatisk hydrolyse af acetamidgruppen. Produktion af den primære metabolit, ucb L057 understøttes ikke af hepatiske cytochrom P450 isoformer. Hydrolyse af acetamidgruppen har været målelig i talrige væv, herunder blodlegemer.Metabolitten ucb L057 er farmakologisk inaktiv.
To mindre metabolitter blev også identificeret. Den ene blev opnået ved hydroxylering af pyrrolidonringen (1,6% af dosis) og den anden fra pyrrolidonringens åbning (0,9% af dosis).
Andre ukendte komponenter tegnede sig kun for 0,6% af dosis.
In vivo der var ingen tegn på enantiomer interkonvertering for hverken levetiracetam eller dets primære metabolit.
In vitro, levetiracetam og dets primære metabolit har vist sig ikke at hæmme aktiviteterne i de store isoformer af humant hepatisk cytochrom P450 (CYP3A4, 2A6, 2C9, 2C19, 2D6, 2E1 og 1A2), glucuronyltransferase (UGT1A1 og UGT1A6) og yderligere epoxidhydroxid , levetiracetam påvirker ikke glucuronidering in vitro af valproinsyre.
I humane hepatocytkulturer havde levetiracetam ringe eller ingen effekt på CYP1A2, SULT1E1 eller UGT1A1. Levetiracetam forårsagede moderat induktion af CYP2B6 og CYP3A4. Dataene in vitro og dataene in vivo relateret til interaktionen med orale præventionsmidler, digoxin og warfarin, indikerer, at der ikke forventes nogen signifikant enzyminduktion in vivo. Derfor er Keppras interaktion med andre stoffer eller den anden vej rundt, det er usandsynligt.
Eliminering
Plasmahalveringstiden hos voksne er 7 ± 1 time og ændrer sig ikke med dosis, indgivelsesvej eller gentagen administration. Gennemsnitlig total kropsclearance er 0,96 ml / min / kg.
Den vigtigste udskillelsesvej er urinvejen, der i gennemsnit er ansvarlig for eliminering af 95% af den administrerede dosis (ca. 93% af dosis udskilles på 48 timer). Fækal eliminering tegner sig kun for 0,3% af dosis.
Den kumulative urinudskillelse af levetiracetam og dets primære metabolit er ansvarlig for eliminering af henholdsvis 66% og 24% af dosis i løbet af de første 48 timer.
Renal clearance af levetiracetam og ucb L057 er henholdsvis 0,6 og 4,2 ml / min / kg, hvilket indikerer, at levetiracetam udskilles ved glomerulær filtrering med efterfølgende tubulær reabsorption, og at den primære metabolit også udskilles ved aktiv tubulær sekretion ud over end med glomerulær filtrering. Elimination af levetiracetam er relateret til kreatininclearance.
Ældre borgere
Hos de "ældre" steg halveringstiden med ca. 40% (10 til 11 timer). Dette skyldes nedsat nyrefunktion i denne population (se pkt.4.2).
Nyresvigt
Den tilsyneladende kropsclearance af både levetiracetam og dets primære metabolit korrelerer med kreatininclearance. Det anbefales derfor at justere den daglige vedligeholdelsesdosis af Keppra baseret på kreatininclearance hos patienter med moderat og svært nedsat nyrefunktion (se pkt. 4.2).
Hos voksne patienter i anurisk nyresygdom i slutstadiet var halveringstiden henholdsvis ca. 25 og 3,1 timer i interdialyse og i dialyseperioder.
Den fjernede levetiracetamfraktion var 51% under en typisk 4-timers dialyse.
Leverinsufficiens
Hos personer med mild til moderat leverinsufficiens var der ingen signifikant ændring af clearance af levetiracetam. Hos de fleste personer med svært nedsat leverfunktion blev clearance af levetiracetam reduceret med mere end 50% på grund af samtidig nyreinsufficiens (se pkt.4.2).
Pædiatrisk population
Børn (fra 4 til 12 år)
Efter en enkelt oral administration (20 mg / kg) til børn (6 til 12 år) med epilepsi var halveringstiden for levetiracetam 6,0 timer. Den tilsyneladende kropsvægt korrigerede clearance var cirka 30% højere end hos voksne med epilepsi.
Efter oral administration af gentagen dosis (20 til 60 mg / kg / dag) til epileptiske børn (4 til 12 år) blev levetiracetam hurtigt absorberet. Højeste plasmakoncentrationer blev observeret 0,5 til 1,0 timer efter dosering. Lineære og dosisproportionelle stigninger blev observeret for maksimale plasmakoncentrationer og areal under kurven.Eliminationshalveringstiden var ca. 5 timer. Tilsyneladende kropsclearance var 1,1 ml / min / kg.
Spædbørn og børn (1 måned til 4 år)
Efter administration af en enkelt dosis (20 mg / kg) 100 mg / ml oral opløsning til epileptiske børn (1 måned til 4 år) blev levetiracetam hurtigt absorberet, og maksimal plasmakoncentration blev observeret ca. 1 time efter administration. Farmakokinetiske resultater viste, at halveringstiden er kortere (5,3 timer) end hos voksne (7,2 timer), og den tilsyneladende clearance var hurtigere (1,5 ml / min / kg) end hos voksne (0, 96 ml / min / kg).
I populationsfarmakokinetiske analyser foretaget hos patienter fra 1 måned til 16 år var kropsvægt signifikant korreleret med tilsyneladende clearance (clearance øget med stigende kropsvægt) og tilsyneladende fordelingsvolumen. Alder påvirkede også. Begge parametre. Denne effekt var markant for de yngre spædbørn og blev svækket med stigende alder for at blive ubetydelig omkring 4 års alderen.
I begge populationsfarmakokinetiske analyser var der en stigning på cirka 20% i den tilsyneladende clearance af levetiracetam ved samtidig administration med et enzyminducerende antiepileptisk lægemiddel.
05.3 Prækliniske sikkerhedsdata
Ikke-kliniske data afslører ingen risiko for mennesker baseret på konventionelle undersøgelser af sikkerhedsfarmakologi, gentoksicitet og kræftfremkaldende potentiale.
Bivirkninger, der ikke blev observeret i kliniske undersøgelser, men set hos rotter og i mindre grad hos mus, ved eksponeringsniveauer svarende til humane eksponeringsniveauer og med mulig relevans for klinisk brug, var leverændringsindekser for respons. Adaptiv, såsom vægtøgning og centrilobulær hypertrofi, fedtinfiltration og forhøjelse af leverenzymer i plasma.
Der blev ikke observeret negative virkninger på han- og hunners fertilitet eller reproduktionsevne hos rotter ved doser op til 1800 mg / kg / dag (6 gange MRHD (Maksimal anbefalet daglig daglig human dosis) baseret på mg / m2 eller baseret på eksponering), både i forældrenes generation og i F1 -generationen.
To embryo-fosterudviklingsstudier (EFD: Embryo-fosterudvikling) blev udført hos rotter ved 400, 1200 og 3600 mg / kg / dag. Ved 3600 mg / kg / dag, i kun en af de 2 EFD -undersøgelser, var der et lille fald i fostervægt forbundet med en marginal stigning i skeletændringer / mindre anomalier. Der var ingen effekt på embryonal dødelighed, og der var heller ikke en stigning i forekomsten af misdannelser. NOAEL (Intet observeret bivirkningsniveau) var 3600 mg / kg / dag for gravide hunrotter (12 gange den maksimalt anbefalede humane daglige dosis (MRHD) baseret på mg / m2) og 1200 mg / kg / dag for fostre.
Fire embryo-fosterudviklingsstudier blev udført på kaniner ved anvendelse af doser på 200, 600, 800, 1200 og 1800 mg / kg / dag. Dosen på 1800 mg / kg / dag inducerede markant toksicitet hos moderen og nedsat fostervægt i forbindelse med en højere forekomst af fostre med kardiovaskulære / skeletmæssige abnormiteter. NOAEL var 2).
En peri- og postnatal udviklingsundersøgelse blev udført hos rotter med levetiracetam-doser på 70, 350, 1800 mg / kg / dag.NOAEL var ≥ 1800 mg / kg / dag for F0 -hunner og for F1 -generationen for overlevelse, vækst og udvikling op til fravænning (6 gange MRHD på mg / m2 -basis).
Undersøgelser med rotter og hunde hos nyfødte og unge dyr har vist, at der ikke forekommer nogen bivirkninger i nogen af standardudviklings- eller modningsmålene ved doser op til 1800 mg / kg / dag (6-17 gange MRHD baseret på mg / m2).
Miljørisikovurdering (Miljørisikovurdering, VAR)
Anvendelse af Keppra i overensstemmelse med oplysningerne i produktresuméet vil sandsynligvis ikke resultere i en uacceptabel miljøpåvirkning (se afsnit 6.6).
06.0 LÆGEMIDDELOPLYSNINGER
06.1 Hjælpestoffer
Kerne:
Croscarmellosenatrium
Macrogol 6000
Vandfri kolloid silica
Magnesiumstearat
Belægning Opadry 85F32004:
Delvist hydrolyseret polyvinylalkohol
Titandioxid (E171)
Macrogol 3350
Talc
Gul jernoxid (E172)
06.2 Uforenelighed
Ikke relevant.
06.3 Gyldighedsperiode
3 år.
06.4 Særlige opbevaringsforhold
Denne medicin kræver ingen særlige opbevaringsbetingelser.
06.5 Den umiddelbare emballages art og emballagens indhold
Aluminium / PVC-blister placeret i papkasser indeholdende 10, 20, 30, 50, 60, 100, 120 filmovertrukne tabletter og multipakninger indeholdende 200 (2 pakninger med 100) filmovertrukne tabletter.
Aluminium / PVC perforerede enhedsdosisblister placeret i papkasser indeholdende 100 x 1 filmovertrukne tabletter.
Ikke alle pakningsstørrelser er nødvendigvis markedsført.
06.6 Brugsanvisning og håndtering
Ubrugt medicin og affald fra denne medicin skal bortskaffes i overensstemmelse med lokale regler.
07.0 INDEHAVER AF MARKEDSFØRINGSTILLADELSE
UCB Pharma SA
Allée de la Recherche 60
B - 1070 Bruxelles
Belgien
08.0 MARKEDSFØRINGSTILLADELSESNUMMER
EU/1/00/146/006 - AIC 035039066
EU/1/00/146/007 - AIC 035039078
EU/1/00/146/008 - AIC 035039080
EU/1/00/146/009 - AIC 035039092
EU/1/00/146/010 - AIC 035039104
EU/1/00/146/011 - AIC 035039116
EU/1/00/146/012 - AIC 035039128
EU/1/00/146/013 - AIC 035039130
EU/1/00/146/035 - AIC 035039332
09.0 DATO FOR FØRSTE TILLADELSE ELLER FORNYELSE AF TILLADELSEN
Dato for første godkendelse: 29. september 2000
Dato for seneste fornyelse: 29. september 2010
10.0 DATO FOR REVISION AF TEKSTEN
August 2013