Aktive ingredienser: Pegfilgrastim
Neulasta 6 mg injektionsvæske, opløsning i en fyldt injektionssprøjte
Indikationer Hvorfor bruges Neulasta? Hvad er det for?
Neulasta indeholder det aktive stof pegfilgrastim. Pegfilgrastim er et protein produceret med en bioteknologisk teknik i en bakteriecelle kaldet Escherichia coli. Det tilhører en gruppe proteiner kaldet cytokiner og ligner meget et naturligt protein (granulocytkolonistimulerende faktor) produceret af vores krop.
Neulasta bruges til at reducere varigheden af neutropeni (lavt antal hvide blodlegemer) og forekomsten af febril neutropeni (lavt antal hvide blodlegemer med feber), som kan skyldes cytotoksisk kemoterapi (medicin, der ødelægger hurtigt voksende celler). Hvide blodlegemer er vigtige, fordi de hjælper kroppen med at bekæmpe infektioner. Disse celler er meget følsomme over for virkningerne af kemoterapi, hvilket kan forårsage et fald i antallet af disse celler i kroppen. Hvis dit antal hvide blodlegemer falder til et lavt niveau, er der muligvis ikke nok tilbage til at bekæmpe bakterierne, og du kan have større risiko for at få en infektion.
Din læge har ordineret Neulasta til at stimulere din knoglemarv (den del af knogle, der danner blodlegemer) til at lave flere hvide blodlegemer til at hjælpe din krop med at bekæmpe infektioner.
Kontraindikationer Når Neulasta ikke bør bruges
Brug ikke Neulasta, hvis du er allergisk over for pegfilgrastim, filgrastim, proteiner afledt af Escherichia coli eller et af de øvrige indholdsstoffer i denne medicin.
Forholdsregler ved brug Hvad du skal vide, før du tager Neulasta
Tal med din læge eller apotek eller sygeplejerske, før du bruger Neulasta:
- hvis du har en allergisk reaktion, herunder svaghed, blodtryksfald, vejrtrækningsbesvær, hævelse i ansigtet (anafylaksi), rødme og rødme, hududslæt og kløende hudområder
- hvis du har en latexallergi. Nålhætten på den fyldte injektionssprøjte indeholder et derivat af latex, som kan forårsage alvorlige allergiske reaktioner
- hvis du har hoste, feber og vejrtrækningsbesvær. Dette kan være et tegn på akut respiratorisk nødsyndrom (ARDS).
- hvis du har en eller flere af følgende bivirkninger:
- hævelse eller hævelse, som kan være forbundet med mindre væskeoverførsel, åndedrætsbesvær, oppustethed i maven og en følelse af fylde og en generel træthedsfornemmelse Disse kan være symptomer på en tilstand kaldet "kapillær lækagesyndrom", der forårsager perfusionblod fra små kar i legeme. Se afsnit 4.
- hvis du har smerter i venstre øvre del af maven eller smerter i skulderens ekstremitet. Disse kan være tegn på miltproblem (splenomegali)
- hvis du for nylig har haft en "alvorlig lungeinfektion (lungebetændelse), væske i lungerne (lungeødem), betændelse i lungerne (interstitiel lungesygdom) eller en" abnormitet fundet ved røntgenstråler (lungeinfiltration)
- hvis du ved, at du har unormale blodlegemer (f.eks. øgede hvide blodlegemer eller anæmi) eller et fald i trombocytniveauer, hvilket reducerer kroppens evne til at størkne (trombocytopeni). Din læge vil måske overvåge dig nøje
- hvis du har seglcelleanæmi. Din læge vil måske overvåge dig nøje
- hvis du pludselig har tegn på allergi såsom udslæt, nældefeber eller kløende hud, hævelse af ansigt, læber, tunge eller andre dele af kroppen, åndenød, hvæsen eller vejrtrækningsbesvær kan dette være tegn på en alvorlig allergisk reaktion.
Din læge vil kontrollere dit blod og urin regelmæssigt, da Neulasta kan beskadige de små filtre inde i dine nyrer (glomerulonephritis).
Du bør tale med din læge om risikoen for at udvikle blodkræft. Hvis du har eller kan have blodkræft, bør du ikke bruge Neulasta, medmindre din læge fortæller dig det.
Tab af reaktion på pegfilgrastim
Hvis du har nedsat respons eller manglende reaktion på behandling med pegfilgrastim, vil din læge undersøge årsagerne, herunder muligheden for at du har udviklet antistoffer, der neutraliserer pegfilgrastims aktivitet.
Interaktioner Hvilke lægemidler eller fødevarer kan ændre virkningen af Neulasta
Fortæl det til din læge eller apotek, hvis du tager anden medicin eller har brugt det for nylig.
Advarsler Det er vigtigt at vide, at:
Graviditet og amning
Spørg din læge eller apotek til råds, før du tager medicin. Neulasta er ikke testet hos gravide. Det er vigtigt at fortælle det til din læge, hvis:
- du er gravid
- mistanke om graviditet eller
- planlægger en graviditet.
Hvis du bliver gravid under behandling med Neulasta, bedes du informere din læge. Du kan blive opfordret til at tilmelde dig Amgens graviditetsovervågningsprogram. Kontaktoplysninger for den lokale repræsentant findes i afsnit 6 i denne indlægsseddel.
Medmindre din læge fortæller dig andet, skal du stoppe med at amme, hvis du bruger Neulasta.
Hvis du ammer, mens du tager Neulasta, kan du blive opfordret til at tilmelde dig Amgens laktationsovervågningsprogram. Kontaktoplysninger til din lokale repræsentant findes i afsnit 6 i denne indlægsseddel.
Kørsel og brug af maskiner
Neulasta har ingen eller ubetydelig indflydelse på evnen til at føre motorkøretøj eller betjene maskiner.
Neulasta indeholder sorbitol (E420) og natriumacetat
Neulasta indeholder sorbitol (en sukkertype). Hvis din læge har fortalt dig, at du ikke tåler visse sukkerarter, skal du kontakte din læge, før du tager denne medicin.
Dette lægemiddel indeholder mindre end 1 mmol (23 mg) natrium pr. 6 mg dosis, det er i det væsentlige natriumfrit.
Dosis, metode og administrationstidspunkt Sådan bruges Neulasta: Dosering
Neulasta er indiceret til voksne i alderen 18 år eller derover.
Tag altid Neulasta nøjagtigt efter lægens anvisning. Hvis du er i tvivl, skal du kontakte din læge eller apotek. Den sædvanlige dosis er en subkutan injektion (injektion under huden) på 6 mg ved hjælp af en fyldt injektionssprøjte, som skal administreres mindst 24 timer efter den sidste dosis kemoterapi ved afslutningen af hver kemoterapicyklus.
Ryst ikke Neulasta kraftigt, da dette kan kompromittere dets aktivitet.
Sådan injiceres dig selv med Neulasta
Din læge kan føle, at det er bedst for dig at injicere Neulasta selv. Din læge eller sygeplejerske vil vise dig, hvordan du injicerer Neulasta. Prøv ikke at injicere dig selv, hvis du ikke har fået at vide, hvordan du injicerer.
Læs afsnittet i slutningen af denne indlægsseddel for instruktioner om, hvordan du selv injicerer Neulasta.
Hvis du har glemt din Neulasta -injektion
Hvis du har glemt en dosis Neulasta, skal du kontakte din læge for at afgøre, hvornår den næste injektion skal tages.
Spørg din læge, apotek eller sygeplejerske, hvis du har yderligere spørgsmål om brugen af dette lægemiddel.
Overdosering Hvad skal man gøre, hvis man har taget for meget Neulasta
Hvis du har brugt for meget Neulasta, skal du kontakte din læge, apotek eller sygeplejerske.
Bivirkninger Hvad er bivirkningerne af Neulasta
Ligesom al anden medicin kan denne medicin forårsage bivirkninger, men ikke alle får bivirkninger.
Fortæl det straks til din læge, hvis du oplever eller en kombination af følgende bivirkninger:
- hævelse eller hævelse, som kan være forbundet med vand, der passerer sjældnere, vejrtrækningsbesvær, oppustethed og følelse af fylde og en generel træthedsfølelse Disse symptomer udvikler sig normalt hurtigt.
Disse kan være symptomer på en usædvanlig tilstand (kan forekomme hos op til 1 ud af 100 mennesker) kaldet "kapillær lækagesyndrom", som får blod til at lække fra små blodkar ind i kroppen og har brug for akut lægehjælp.
Meget almindelige bivirkninger (som kan påvirke mere end 1 ud af 10 personer):
- knoglesmerter. Din læge vil fortælle dig, hvad du skal tage for at lindre knoglesmerter.
- kvalme og hovedpine.
Almindelige bivirkninger (som kan forekomme hos op til 1 ud af 10 personer):
- smerter på injektionsstedet.
- generelle smerter i led og muskler.
- nogle ændringer kan forekomme i blodet, men disse vil blive opdaget under rutinemæssige blodprøver. Niveauerne af hvide blodlegemer kan stige i en kort periode. Trombocyttalniveauerne kan falde og forårsage blå mærker.
Ikke almindelige bivirkninger (som kan forekomme hos op til 1 ud af 100 personer):
- allergiske reaktioner, herunder rødme og rødme, hududslæt (rødme i huden) og kløende hævelse af huden.
- alvorlige allergiske reaktioner, herunder anafylaksi (svaghed, blodtryksfald, åndedrætsbesvær, hævelse i ansigtet).
- stigning i miltens volumen.
- brud på milten. Nogle tilfælde af bristet milt har været dødelige. Det er vigtigt, at du straks kontakter din læge, hvis du føler smerter i øverste venstre side af maven eller venstre skulder, da dette kan indikere problemer med milten.
- vejrtrækningsproblemer. Kontakt din læge, hvis du har hoste, feber og vejrtrækningsbesvær.
- der har været tilfælde af Sweet's syndrom (lilla, hævede og smertefulde læsioner på lemmerne og undertiden i ansigt og hals, forbundet med feber), men andre faktorer kan have bidraget
- kutan vaskulitis (betændelse i hudens blodkar).
- skade på de små filtre inde i nyrerne (glomerulonephritis).
- rødme på injektionsstedet.
Indberetning af bivirkninger
Tal med din læge eller apotek eller sygeplejerske, hvis du får bivirkninger. Dette inkluderer eventuelle bivirkninger, der ikke er anført i denne indlægsseddel. Du kan også rapportere bivirkninger direkte via det nationale rapporteringssystem anført i tillæg V.
Udløb og opbevaring
Opbevar denne medicin utilgængeligt for børn.
Brug ikke dette lægemiddel efter den udløbsdato, der står på kartonen og sprøjtemærket efter EXP. Udløbsdatoen refererer til den sidste dag i den pågældende måned.
Opbevares i køleskab (2 ° C - 8 ° C).
Du kan tage Neulasta ud af køleskabet og opbevare det ved stuetemperatur (højst 30 ° C) i højst 3 dage. Når sprøjten er fjernet fra køleskabet og har nået stuetemperatur (ikke over 30 ° C), skal den bruges inden for 3 dage eller smides væk.
Må ikke fryses. Neulasta kan bruges, hvis det ved et uheld har været frosset én gang i mindre end 24 timer.
Opbevar beholderen i den ydre karton for at beskytte medicinen mod lys.
Brug ikke dette lægemiddel, hvis du bemærker, at det er grumset, eller du ser partikler.
Smid ikke medicin via spildevand eller husholdningsaffald. Spørg din apotek om, hvordan du skal smide medicin, du ikke længere bruger. Dette vil hjælpe med at beskytte miljøet.
Deadline "> Andre oplysninger
Neulasta indeholder
- Den aktive ingrediens er pegfilgrastim. Hver fyldt injektionssprøjte indeholder 6 mg pegfilgrastim i 0,6 ml opløsning.
- Øvrige indholdsstoffer er natriumacetat, sorbitol (E420), polysorbat 20 og vand til injektionsvæsker. Se afsnit 2.
Hvordan Neulasta ser ud og pakningens indhold
Neulasta er en klar, farveløs injektionsvæske, opløsning i en fyldt injektionssprøjte (6 mg / 0,6 ml).
Hver pakning indeholder 1 fyldt injektionssprøjte af glas med nål og kanylehætte i rustfrit stål. Sprøjterne er pakket med blister eller uden blister.
Udløb "> Instruktioner til injektion af Neulasta fyldt injektionssprøjte
Dette afsnit indeholder oplysninger om, hvordan du selv injicerer Neulasta.
Det er vigtigt, at du ikke forsøger at injicere dig selv, hvis du ikke har fået at vide, hvordan du gør det af din læge, sygeplejerske eller apotek. Spørg din læge, sygeplejerske eller apoteket for at få hjælp, hvis du har spørgsmål om, hvordan du injicerer.
Sådan bruges, af dig eller af personen, der giver dig injektionen, af Neulasta i en fyldt injektionssprøjte
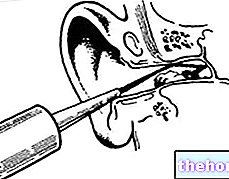
Du bliver nødt til at injicere dig selv lige under huden Denne injektion kaldes subkutan.
Hvad skal der til
For at give dig selv en "subkutan injektion skal du bruge:
- en fyldt injektionssprøjte med Neulasta; Og
- alkoholservietter eller lignende desinfektionsmidler.
Hvad skal jeg gøre, før jeg giver mig selv en "subkutan injektion af Neulasta?
- Tag medicinen ud af køleskabet.
- Ryst ikke den fyldte sprøjte.
- Fjern ikke kanylehætten fra sprøjten, før du er klar til at injicere.
- Kontroller udløbsdatoen på etiketten på den fyldte sprøjte (EXP). Brug den ikke efter den sidste dag i den viste måned.
- Kontroller udseendet af Neulasta. Det skal være en klar, farveløs væske. Hvis du ser partikler, bør du ikke bruge det.
- For en mere behagelig injektion, lad den fyldte sprøjte stå uden for køleskabet i en halv time for at nå stuetemperatur eller hold den forsigtigt i hånden i et par minutter. Opvarm ikke Neulasta på anden måde (f.eks. Opvarm den ikke i en mikrobølgeovn eller varmt vand).
- Vask dine hænder grundigt.
- Find en behagelig, godt oplyst og ren overflade, og hav alt, hvad du har brug for, lige ved hånden.
Hvordan forbereder jeg injektionen af Neulasta?
Inden du giver dig selv injektionen af Neulasta, skal du gøre følgende:
- Tag sprøjten i din hånd, og fjern forsigtigt hætten fra nålen uden at bøje den. Træk vandret. Rør ikke ved nålen eller skub stemplet.
- Du vil muligvis bemærke en lille luftboble i den fyldte sprøjte. Du må ikke fjerne luftboblen før injektion. Injektion af opløsningen med luftboblen er ufarlig.
- Du kan nu bruge den fyldte sprøjte.
Hvor skal jeg give mig selv injektionen?
De mest egnede steder at injicere dig selv er:
- den øverste del af lårene; Og
- maven, undtagen området omkring navlen.
Hvis en anden giver dig injektionen, kan du også bruge bagsiden af dine arme.
Hvordan giver jeg mig selv injektionen?
- Rengør din hud med en alkoholserviet.
- Løft huden mellem tommelfinger og pegefinger (uden at klemme den). Skub nålen ind i din hud.
- Skub stemplet ned med langsomt, stabilt tryk. Skub stemplet helt ind, indtil al væsken er sprøjtet ind.
- Efter at have injiceret væsken, trækkes nålen ud og slipper huden.
- Hvis du bemærker en lille dråbe blod på injektionsstedet, skal du forsigtigt tørre det af med en vatpind eller gaze. Gnid ikke injektionsstedet. Om nødvendigt kan du dække injektionsstedet med et plaster.
- Genbrug ikke den resterende Neulasta i sprøjten.
At huske
Brug kun hver sprøjte til én injektion. Hvis du har problemer, tøv ikke med at kontakte din læge eller sygeplejerske for at få hjælp og råd.
Bortskaffelse af brugte sprøjter
- Sæt ikke hætten tilbage på brugte nåle.
- Opbevar brugte sprøjter utilgængeligt for børn.
- Brugte sprøjter skal bortskaffes i overensstemmelse med lokale krav. Spørg din apotek om, hvordan du skal smide medicin, du ikke længere bruger. Dette vil hjælpe med at beskytte miljøet.
Indlægsseddel: AIFA (Italian Medicines Agency). Indhold offentliggjort i januar 2016. De foreliggende oplysninger er muligvis ikke opdaterede.
For at få adgang til den mest opdaterede version er det tilrådeligt at få adgang til webstedet AIFA (Italian Medicines Agency). Ansvarsfraskrivelse og nyttige oplysninger.
01.0 LÆGEMIDLETS NAVN -
NEULASTA 6 MG LØSNING TIL INJEKTION
02.0 KVALITATIV OG KVANTITATIV SAMMENSÆTNING -
Hver fyldt injektionssprøjte indeholder 6 mg pegfilgrastim * i 0,6 ml injektionsvæske, opløsning. Koncentrationen er 10 mg / ml i betragtning af kun proteindelen **.
* Pegfilgrastim produceres i celler af Escherichia coli med rekombinant DNA -teknologi og efterfølgende konjugering med polyethylenglycol (PEG).
** Koncentrationen er 20 mg / ml, hvis PEG -delen af molekylet er inkluderet.
Dette produkts styrke bør ikke sammenlignes med virkningen af andre pegylerede eller ikke-pegylerede proteiner, der tilhører samme terapeutiske klasse.
For mere information, se afsnit 5.1.
Hjælpestof (er) med kendt virkning:
Hver fyldt injektionssprøjte indeholder 30 mg sorbitol (E420)
Hver fyldt injektionssprøjte indeholder mindre end 1 mmol (23 mg) natrium (se pkt. 4.4).
Den fulde liste over hjælpestoffer findes i afsnit 6.1.
03.0 LÆGEMIDDELFORM -
Injicerbar løsning.
Klar og farveløs injektionsvæske, opløsning.
04.0 KLINISKE OPLYSNINGER -
04.1 Terapeutiske indikationer -
Reduktion i varigheden af neutropeni og forekomsten af febril neutropeni hos voksne patienter behandlet med cytotoksisk kemoterapi mod kræft (med undtagelse af kronisk myeloid leukæmi og myelodysplastiske syndromer).
04.2 Dosering og indgivelsesmåde -
Neulasta -behandling bør initieres og overvåges af læger med erfaring inden for onkologi og / eller hæmatologi.
Dosering
En dosis på 6 mg (en enkelt fyldt injektionssprøjte) Neulasta anbefales for hver kemoterapicyklus, administreret mindst 24 timer efter cytotoksisk kemoterapi.
Indgivelsesmåde
Neulasta injiceres subkutant. Injektionen skal gives i låret, maven eller overarmen. For instruktioner om håndtering af lægemidlet før administration, se afsnit 6.6.
Pædiatrisk population
Sikkerhed og virkning af Neulasta hos børn er endnu ikke fastslået. Aktuelt tilgængelige data er beskrevet i afsnit 4.8, 5.1 og 5.2, men der kan ikke fremsættes anbefalinger om dosering.
Patienter med nedsat nyrefunktion
Det anbefales ikke at justere dosis til patienter med nedsat nyrefunktion, herunder patienter med nyresygdom i slutstadiet.
04.3 Kontraindikationer -
Overfølsomhed over for det aktive stof eller over for et eller flere af hjælpestofferne.
04.4 Særlige advarsler og passende forholdsregler ved brug -
Begrænsede kliniske data tyder på en sammenlignelig effekt af pegfilgrastim sammenlignet med filgrastim til tiden til remission fra alvorlig neutropeni hos patienter med akut myeloid leukæmi. de novo (se afsnit 5.1). Langtidseffekterne af Neulasta ved akut myeloid leukæmi er imidlertid ikke blevet fastslået; derfor bør produktet bruges med forsigtighed i denne patientpopulation.
Den granulocytkolonistimulerende faktor kan fremme væksten af myeloide celler in vitro og lignende effekter kan observeres in vitro i nogle ikke-myeloide celler.
Sikkerheden og effekten af Neulasta er ikke undersøgt hos patienter med myelodysplastisk syndrom, kronisk myeloid leukæmi og hos patienter med sekundær akut myeloid leukæmi (AML); derfor bør det ikke anvendes til sådanne patienter. Der bør udvises særlig omhu for at skelne mellem diagnose af eksplosionstransformation af kronisk myeloid leukæmi fra akut myeloid leukæmi.
Effekten og sikkerheden ved administration af Neulasta til patienter med AML de novo af alder
Sikkerhed og virkning af Neulasta hos patienter, der får kemoterapi i høj dosis, er ikke undersøgt.Dette lægemiddel bør ikke bruges til at øge doserne af cytotoksisk kemoterapi ud over standarddosisregimer.
Lungebivirkninger
Ikke almindelige lungebivirkninger (≥ 1 / 1.000, interstitiel lungebetændelse er blevet rapporteret efter administration af G-CSF. Patienter med en nylig historie med lungeinfiltrater eller lungebetændelse kan have større risiko (se pkt. 4.8).
Indtræden af lungesymptomer som hoste, feber og dyspnø på samme tid som et radiologisk billede af lungeinfiltrater og en forringelse af lungefunktionen forbundet med et forhøjet antal hvide blodlegemer kan være de første tegn på akut respiratorisk nødsyndrom (Acute Respiratory Distress syndrom, ARDS). Under sådanne omstændigheder bør Neulasta -behandlingen, efter lægens skøn, afbrydes og passende behandling indledes (se pkt. 4.8).
Glomerulonephritis
Glomerulonephritis er blevet rapporteret hos patienter, der fik filgrastim og pegfilgrastim. Generelt forsvandt glomerolunephritis -hændelser efter dosisreduktion eller seponering af filgrastim og pegfilgrastim. Overvågning af urinalyse anbefales.
Kapillær lækagesyndrom
Kapillær lækagesyndrom er blevet rapporteret efter administration af granulocytkolonistimulerende faktorer og er karakteriseret ved hypotension, hypoalbuminæmi, ødem og hæmokoncentration. Patienter, der udvikler symptomer på kapillær lækagesyndrom, bør overvåges nøje og modtage standard symptomatisk behandling, hvilket kan omfatte behovet for intensiv pleje (se pkt. 4.8).
Splenomegali og miltbrud
Usædvanlige, men generelt asymptomatiske tilfælde af splenomegali og usædvanlige tilfælde af miltbrud, herunder nogle dødelige tilfælde, er blevet rapporteret efter administration af pegfilgrastim (se pkt. 4.8). Derfor skal miltens volumen overvåges omhyggeligt (f.eks. Ved klinisk undersøgelse, ultralyd). En diagnose af miltbrud bør overvejes hos patienter, der har smerter i mave- eller skulder i venstre øvre kvadrant.
Trombocytopeni og anæmi
Behandling med Neulasta alene udelukker ikke trombocytopeni og anæmi forårsaget af opretholdelse af fulde doser af myelosuppressiv kemoterapi som planlagt. Regelmæssig monitorering af trombocyttal og hæmatokrit anbefales. Der bør lægges særlig vægt på administration af enkelt- eller kombinationskemoterapeutiske midler, der forårsager alvorlig trombocytopeni.
Seglcelleanæmi
Seglcellekriser har været forbundet med brug af pegfilgrastim til patienter med seglcelleegenskab eller med seglcellesygdom (se pkt. 4.8). Derfor bør lægen udvise forsigtighed ved ordination af Neulasta til patienter med seglcelleegenskab eller seglcellesygdom. behold passende kliniske og laboratorieparametre, og du skal være opmærksom på den mulige sammenhæng mellem denne medicin og en forstørret milt og en vaso-okklusiv krise.
Leukocytose
Værdier for hvide blodlegemer (Hvide blodlegemer, WBC) svarende til eller større end 100 x 109 / l er blevet observeret hos mindre end 1% af patienterne behandlet med Neulasta Ingen bivirkninger, der direkte kan tilskrives denne grad af leukocytose, er blevet rapporteret. Denne stigning i antallet af hvide blodlegemer er forbigående , observeres typisk 24 - 48 timer efter administration og er i overensstemmelse med lægemidlets farmakodynamiske virkninger I overensstemmelse med de kliniske virkninger og muligheden for leukocytose bør der udføres et hvidt blodlegemer (WBC) med jævne mellemrum under behandlingen Hvis antallet af hvide blodlegemer overstiger 50 x 109 / L efter den forventede nadir, administration af dette lægemiddel bør straks stoppes.
Overfølsomhed
Overfølsomhedsreaktioner, herunder anafylaktiske reaktioner, der forekommer ved initiering eller efter behandling, er blevet rapporteret hos patienter behandlet med Neulasta Afbryd behandlingen med Neulasta permanent hos patienter med klinisk signifikant overfølsomhed. Administrer ikke Neulasta til patienter med en historie med overfølsomhed over for pegfilgrastim eller filgrastim .. Hvis der opstår en alvorlig allergisk reaktion, bør passende behandling gives, efterfulgt af omhyggelig opfølgning af patienten i flere dage.
Immunogenicitet
Som med alle terapeutiske proteiner er der en potentiel risiko for immunogenicitet. Sandsynligheden for at generere antistoffer mod pegfilgrastim er generelt lav. Udvikling af bindende antistoffer forventes med alle biologiske stoffer, men hidtil har de ikke været forbundet med aktivitet. Neutralisering.
Sikkerheden og effekten af Neulasta ved hæmatopoietisk stamcellemobilisering hos raske patienter eller donorer er ikke blevet vurderet tilstrækkeligt.
Nålehætten på den fyldte sprøjte indeholder tørt naturgummi (et derivat af latex), som kan forårsage allergiske reaktioner.
Øget hæmatopoietisk aktivitet af knoglemarven som reaktion på vækstfaktorterapi har været forbundet med forbigående positive knogleradiologiske fund.Dette bør overvejes ved fortolkning af radiologiske data.
Neulasta indeholder sorbitol. Patienter med sjældne arvelige problemer med fructoseintolerance bør ikke tage denne medicin.
Neulasta indeholder mindre end 1 mmol (23 mg) natrium i en dosis på 6 mg, dvs. det er i det væsentlige "natriumfrit".
For at forbedre sporbarheden af granulocytkolonistimulerende faktorer (G-CSF) bør handelsnavnet for det administrerede produkt tydeligt registreres i patientjournalen.
04.5 Interaktioner med andre lægemidler og andre former for interaktion -
I betragtning af den potentielle følsomhed for hurtigt at dele myeloide celler til cytotoksisk kemoterapi, bør Neulasta administreres mindst 24 timer efter administration af cytotoksisk kemoterapi. I kliniske undersøgelser viste administration af Neulasta 14 dage før kemoterapi at være sikker. Brugen af Neulasta samtidig med kemoterapi er ikke blevet evalueret hos patienter.I dyremodeller har samtidig administration af Neulasta og 5-fluorouracil (5-FU) eller andre antimetabolitter vist sig at forværre myelosuppression..
Kliniske undersøgelser undersøgte ikke specifikt mulige interaktioner med andre hæmatopoietiske vækstfaktorer og cytokiner.
Den potentielle interaktion med lithium, som også fremmer frigivelse af neutrofiler, er ikke specifikt undersøgt.Der er ingen tegn på, at denne interaktion kan være skadelig.
Sikkerheden og effekten af Neulasta er ikke blevet evalueret hos patienter, der får kemoterapi i forbindelse med forsinket myelosuppression, såsom nitrosourea.
Der er ikke udført specifikke undersøgelser af interaktioner eller stofskifte; kliniske undersøgelser har imidlertid ikke vist nogen interaktioner mellem Neulasta og andre lægemidler.
04.6 Graviditet og amning -
Graviditet
Der er ingen eller begrænsede data om brugen af pegfilgrastim til gravide Dyrestudier har vist reproduktionstoksicitet (se pkt. 5.3) Neulasta anbefales ikke under graviditet og hos kvinder i den fertile alder, der ikke anvender prævention.
Kvinder, der viser sig at være gravide under behandling med Neulasta, opfordres til at tilmelde sig Amgens graviditetsovervågningsprogram. Kontaktoplysninger findes i afsnit 6 i indlægssedlen.
Fodringstid
Der er utilstrækkelige oplysninger om udskillelse af Neulasta / metabolitter i modermælk. En risiko for de nyfødte / spædbørn kan ikke udelukkes. Der skal træffes beslutning om, hvorvidt amning skal ophøre eller om at afbryde / afstå fra Neulasta-behandling under hensyntagen til fordelen ved amning til barnet og fordelen ved terapi for kvinden.
Ammende kvinder under behandling med Neulasta opfordres til at tilmelde sig Amgens laktationsovervågningsprogram. Kontaktoplysninger findes i afsnit 6 i indlægssedlen.
Fertilitet
Pegfilgrastim havde ingen effekt på reproduktionsevne eller fertilitet hos han- eller hunrotter ved den kumulative ugentlige dosis på cirka 6 til 9 gange den højeste anbefalede humane dosis (baseret på kropsoverfladeareal) (se pkt. 5.3).
04.7 Virkninger på evnen til at føre motorkøretøj og betjene maskiner -
Neulasta har ingen eller ubetydelig indflydelse på evnen til at føre motorkøretøj eller betjene maskiner.
04.8 Bivirkninger -
Resumé af sikkerhedsprofilen
De hyppigst rapporterede bivirkninger var knoglesmerter (meget almindelige [≥ 1/10]) og muskuloskeletale smerter (almindelige). Knoglesmerter var generelt milde til moderate, forbigående og kunne kontrolleres med almindelige smertestillende midler hos de fleste patienter.
Tilfælde af overfølsomhedsreaktioner, herunder hududslæt, urticaria, angioødem, dyspnø, erytem, rødme og hypotension, er blevet rapporteret ved den første eller efterfølgende administration af Neulasta (usædvanlig [≥ 1 / 1.000, anafylaksi, kan forekomme hos patienter, der får Neulasta (ikke almindelig ) (se afsnit 4.4).
Kapillær lækagesyndrom, som kan være livstruende, hvis behandlingen forsinkes, er blevet rapporteret som usædvanlig (≥ 1 / 1.000 til
Splenomegali, normalt asymptomatisk, er ualmindelig.
Usædvanlige tilfælde af miltbrud, herunder nogle dødelige tilfælde, er blevet rapporteret efter administration af pegfilgrastim (se pkt. 4.4).
Ikke almindelige lungebivirkninger, herunder interstitiel lungebetændelse, lungeødem, lungeinfiltrater og lungefibrose er blevet rapporteret. Usædvanlige tilfælde har resulteret i respirationssvigt eller akut respiratorisk nødsyndrom (Acute Respiratory Distress syndrom, ARDS), som kan være dødelig (se pkt. 4.4).
Isolerede tilfælde af seglcellekriser (ualmindeligt hos sådanne patienter) er blevet rapporteret hos patienter med seglstræk eller seglcellesygdom (se pkt. 4.4).
Tabel over bivirkninger
Dataene i nedenstående tabel beskriver de bivirkninger, der er rapporteret i kliniske undersøgelser og spontane rapporter. Inden for hver frekvensklasse rapporteres bivirkninger i faldende sværhedsgrad.
¹ Se afsnittet "Beskrivelse af udvalgte bivirkninger" nedenfor.
² Denne bivirkning blev identificeret gennem overvågning efter markedsføring, men blev ikke observeret i randomiserede kontrollerede forsøg med voksne. Frekvensklassen blev bestemt med en statistisk beregning baseret på 1.576 patienter behandlet med Neulasta i ni randomiserede kliniske forsøg.
Beskrivelse af udvalgte bivirkninger
Usædvanlige tilfælde af Sweet's syndrom er blevet rapporteret, selvom den underliggende tilstedeværelse af hæmatologiske maligniteter i nogle tilfælde kan have bidraget.
Usædvanlige hændelser med kutan vaskulitis er blevet rapporteret hos patienter behandlet med Neulasta. Mekanismen, der forårsager vaskulitis hos patienter behandlet med Neulasta, er ukendt.
Reaktioner på injektionsstedet, herunder erytem på injektionsstedet (ikke almindelig (≥ 1 / 1.000,
Almindelige tilfælde (≥ 1/100, 100 x 109 / l) er blevet rapporteret (se pkt. 4.4).
Hos patienter behandlet med Neulasta efter cytotoksisk kemoterapi er reversible, milde eller moderate forhøjelser, ikke ledsaget af kliniske symptomer, i urinsyre og alkalisk phosphatase ualmindelige; Reversible, milde eller moderate forhøjelser, der ikke ledsages af kliniske symptomer, i laktatdehydrogenase er ualmindelige.
Kvalme og hovedpine blev observeret meget almindeligt hos patienter i kemoterapi.
Usædvanlige tilfælde af forhøjede leverfunktionstests (LFT) for ALAT (alaninaminotransferase) eller ASAT (aspartataminotransferase) er blevet observeret hos patienter, der fik pegfilgrastim efter cytotoksisk kemoterapi. Disse stigninger er forbigående og reversible.
Almindelige tilfælde af trombocytopeni er blevet rapporteret.
Tilfælde af kapillær lækagesyndrom er blevet rapporteret efter markedsføring med brug af granulocytkolonistimulerende faktorer Disse er generelt forekommet hos patienter med fremskreden malign sygdom, sepsis, der tager flere kemoterapilægemidler eller er under aferes (se pkt. 4.4).
Pædiatrisk population
Erfaring med børn er begrænset. En højere hyppighed af alvorlige bivirkninger blev observeret hos børn i alderen 0-5 år (92%) sammenlignet med ældre børn i henholdsvis 6-11 og 12-21 år (80%og 67%) og voksne. Den hyppigste rapporterede bivirkning var knoglesmerter (se pkt. 5.1 og 5.2).
Indberetning af formodede bivirkninger
Rapportering af formodede bivirkninger, der opstår efter godkendelse af lægemidlet, er vigtig, da det muliggør kontinuerlig overvågning af lægemidlets fordel / risiko -balance.Professionelle sundhedspersonale anmodes om at rapportere alle formodede bivirkninger via det nationale rapporteringssystem (Italian Medicines Agency - Websted: www.agenziafarmaco.gov.it/it/responsabili).
04.9 Overdosering -
En enkelt dosis på 300 mcg / kg blev administreret subkutant til et begrænset antal raske frivillige og til patienter med non-microcytoma lungekræft uden alvorlige bivirkninger. Bivirkninger lignede dem hos forsøgspersoner, der fik lavere doser pegfilgrastim.
05.0 FARMAKOLOGISKE EGENSKABER -
05.1 "Farmakodynamiske egenskaber -
Farmakoterapeutisk gruppe: immunstimulerende midler, kolonistimulerende faktor.
ATC -kode: L03AA13.
Human granulocytkolonistimulerende faktor (G-CSF) er et glycoprotein, der regulerer produktion og frigivelse af neutrofiler fra knoglemarven. Pegfilgrastim består af et rekombinant humant G-CSF-molekyle (r-metHuG-CSF) kovalent bundet til et enkelt 20 kd polyethylenglycol (PEG) molekyle. Pegfilgrastim er en langvarig form for filgrastim på grund af reduceret renal clearance. Pegfilgrastim og filgrastim har identiske virkningsmekanismer, hvilket forårsager en markant stigning i antallet af perifere neutrofiler inden for 24 timer med ubetydelige stigninger i monocytter og / eller lymfocytter. Ligesom filgrastim udviser neutrofiler produceret som reaktion på pegfilgrastim normal eller øget funktion, som demonstreret ved vurderinger af kemotaktisk og fagocytisk aktivitet.Ligesom andre hæmatopoietiske vækstfaktorer har G-CSF vist in vitro stimulerende egenskaber på humane endotelceller. G-CSF kan fremme vækst in vitro af myeloide celler kan endda maligne og lignende effekter påvises in vitro på nogle ikke-myeloide celler.
I to randomiserede, dobbeltblinde, pivotale undersøgelser af patienter med højrisiko II-IV-brystkræft i fase II, der gennemgik myelosuppressiv kemoterapi, herunder doxorubicin og docetaxel, reducerede brugen af pegfilgrastim som en enkelt dosis én gang pr. Cyklus varigheden af neutropeni og forekomsten af febril neutropeni svarende til den, der blev observeret ved daglig dosering af filgrastim (median på 11 doseringsdage). I mangel af vækstfaktorunderstøttelse er der rapporteret om, at dette mønster resulterer i en gennemsnitlig varighed af grad 4 neutropeni på 5-7 dage med en forekomst af febril neutropeni på 30-40%. I en undersøgelse (n = 157) med en fast dosis på 6 mg pegfilgrastim, den gennemsnitlige varighed af grad 4 neutropeni for pegfilgrastim -gruppen var 1,8 dage sammenlignet med 1,6 dage i filgrastim -gruppen (forskel 0,23 dage, 95% CI: -0,15, 0,63). Under hele undersøgelsen , febern neutropeni var 13% af patienterne behandlet med pegfilgrastim sammenlignet med 20% af patienterne behandlet med filgrastim (forskel 7%, 95% CI: - 19%, 5%). I en anden undersøgelse (n = 310) ved hjælp af en vægtjusteret dosis (100 mcg / kg) var den gennemsnitlige varighed af grad 4 neutropeni i pegfilgrastim-gruppen 1,7 dage sammenlignet med 1,8 dage i filgrastim-gruppen. (Forskel 0,03 dage, 95% CI: -0,36, 0,30). Den samlede rate af febril neutropeni var 9% af patienterne behandlet med pegfilgrastim og 18% af patienterne behandlet med filgrastim (forskel 9%, 95% CI: -16,8%, -1,1%).
I et dobbeltblindet, placebokontrolleret studie med brystkræftpatienter blev effekten af pegfilgrastim på forekomsten af febril neutropeni evalueret efter administration af et kemoterapiregime forbundet med en 10-20% forekomst af febril neutropeni (docetaxel 100 mg / m² hver 3. uge i 4 cyklusser). Ni hundrede og tyve patienter blev randomiseret til at modtage en enkelt dosis pegfilgrastim eller placebo cirka 24 timer efter kemoterapi i hver cyklus (dag 2.) Forekomsten af febril neutropeni var lavere hos randomiserede patienter. til modtage pegfilgrastim versus placebo (1% versus 17%, s
En begrænset prøve (n = 83) Fase II, randomiseret, dobbeltblind undersøgelse udført hos patienter, der gennemgår kemoterapi for akut myeloid leukæmi de novo sammenlignet pegfilgrastim (enkeltdosis på 6 mg) med filgrastim givet under induktionskemoterapi. Mediantiden til remission fra alvorlig neutropeni var 22 dage i begge behandlingsgrupper. Det langsigtede resultat er ikke undersøgt (se pkt. 4.4).
I et multicenter, randomiseret, åbent fase II-studie (n = 37) hos pædiatriske patienter med sarkom, der modtog 100 mcg / kg pegfilgrastim efter det første kursus af kemoterapi med vincristin, doxorubicin og cyclophosphamid (VAdriaC / IE), en længere varighed af alvorlig neutropeni (neutrofiler
05.2 "Farmakokinetiske egenskaber -
Den maksimale serumkoncentration af pegfilgrastim observeres 16 til 120 timer efter administration af en enkelt subkutan dosis; serumkoncentrationer forbliver stabile i perioden med neutropeni efter myelosuppressiv kemoterapi. Elimination af pegfilgrastim er ikke-lineær med hensyn til dosis; serumclearance af pegfilgrastim falder med stigende dosis. Pegfilgrastim synes hovedsageligt at blive elimineret gennem neutrofil-medieret clearance, som er mættet ved højere doser. I overensstemmelse med en selvreguleret clearancemekanisme falder serumkoncentrationen af pegfilgrastim hurtigt i forbindelse med stigningen af neutrofiler.
På grund af den neutrofil-medierede clearance-mekanisme forventes nedsat lever- eller nyrefunktion ikke at påvirke farmakokinetikken af pegfilgrastim. I et åbent enkeltdosisundersøgelse (n = 31) påvirkede forskellige stadier af nedsat nyrefunktion, herunder nyresygdom i slutstadiet, ikke pegfilgrastims farmakokinetik.
Ældre befolkning
De begrænsede tilgængelige data indikerer, at pegfilgrastims farmakokinetik hos ældre personer (> 65 år) ligner den hos voksne.
Pædiatrisk population
Farmakokinetikken for pegfilgrastim blev undersøgt hos 37 pædiatriske sarkom -patienter, der modtog 100 μg / kg pegfilgrastim efter afslutning af VAdriaC / IE kemoterapi. Den yngre aldersgruppe (0-5 år) havde højere "gennemsnitlig pegfilgrastim-eksponering (AUC) (± standardafvigelse) (47,9 ± 22,5 mcg • time / ml) end børn ældre end 6-11 år og 12-21 år (22,0 ± 13,1 mcg • hr / ml og henholdsvis 29,3 ± 23,2 mcg • hr / ml) (se afsnit 5.1).
Med undtagelse af den yngre aldersgruppe (0-5 år) så den gennemsnitlige AUC hos pædiatriske patienter ud som for voksne patienter med fase II-IV højrisiko brystkræft, der modtog 100 mcg / kg pegfilgrastim efter afslutning af doxorubicin / docetaxel (se afsnit 4.8 og 5.1).
05.3 Prækliniske sikkerhedsdata -
Prækliniske data fra traditionelle toksicitetsundersøgelser ved gentagen dosis afslørede forventede farmakologiske virkninger, herunder stigninger i antallet af hvide blodlegemer, knoglemarvs myeloid hyperplasi, ekstramedullær hæmatopoiesis og splenomegali.
Der blev ikke observeret nogen bivirkninger hos rotter født til gravide kvinder, hvortil pegfilgrastim blev administreret subkutant, men hos kaniner forårsagede pegfilgrastim administreret subkutant embryo-føtal toksicitet (tab af embryoet) ved kumulative doser på 4 gange den anbefalede dosis til mennesker. Undersøgelser på rotter har vist, at transplacental passage af pegfilgrastim er mulig. Undersøgelser med rotter viste, at subkutan administration af pegfilgrastim ikke havde nogen effekt på reproduktionsevne, fertilitet, brunstcyklus, dage mellem parring og coitus og intrauterin overlevelse. Relevansen af disse data for mennesker er ukendt.
06.0 LÆGEMIDDELOPLYSNINGER -
06.1 Hjælpestoffer -
Natriumacetat *
Sorbitol (E420)
Polysorbat 20
Vand til injektionsvæsker
* Natriumacetat opnås ved titrering af iseddike med natriumhydroxid.
06.2 Uforenelighed "-
Dette lægemiddel må ikke blandes med andre produkter, især natriumchloridopløsninger.
06.3 Gyldighedsperiode "-
3 år.
06.4 Særlige opbevaringsforhold -
Opbevares i køleskab (2 ° C - 8 ° C).
Neulasta kan opbevares ved stuetemperatur (ikke over 30 ° C) én gang og i en maksimal periode på 72 timer. Neulasta, der efterlades ved stuetemperatur i mere end 72 timer, skal kasseres.
Må ikke fryses. Utilsigtet udsættelse for kuldegrader, én gang i mindre end 24 timer, påvirker ikke Neulastas stabilitet.
Opbevar beholderen i den ydre karton for at beskytte medicinen mod lys.
06.5 Den umiddelbare emballages art og emballagens indhold -
Forfyldt sprøjte (type I-glas) med gummistempel og rustfri nål med eller uden automatisk nålebeskytter.
Nålehætten på den fyldte sprøjte indeholder tørt naturgummi (et derivat af latex) (se pkt.4.4).
Hver fyldt injektionssprøjte indeholder 0,6 ml injektionsvæske, opløsning. Pakningsstørrelse på en fyldt injektionssprøjte pakket med blister eller uden blister.
Ikke alle pakningsstørrelser er nødvendigvis markedsført.
06.6 Brugsanvisning og håndtering -
Inden administration af Neulasta -opløsningen skal fraværet af synlige partikler kontrolleres. Kun en klar og farveløs opløsning skal injiceres.
Ved overdreven omrøring kan pegfilgrastim danne aggregater og blive biologisk inaktive.
Lad den fyldte sprøjte nå stuetemperatur, inden opløsningen injiceres.
Ubrugt medicin og affald fra denne medicin skal bortskaffes i overensstemmelse med lokale regler.
07.0 INDEHAVER AF "MARKEDSFØRINGSTILLADELSEN" -
Amgen Europe B.V.
Minervum 7061
4817 ZK Breda
Holland
08.0 MARKEDSFØRINGSTILLADELSESNUMMER -
EU/1/02/227/001 1 pakningssprøjte med blister
035716012
EU/1/02/227/002 1 sprøjtepakning uden blister
EU/1/02/227/004 1 -pakningssprøjte med blister med nålebeskytter
035716036
09.0 DATO FOR FØRSTE TILLADELSE ELLER FORNYELSE AF TILLADELSEN -
Dato for første godkendelse: 22. august 2002
Dato for seneste fornyelse: 16. juli 2007
10.0 DATO FOR REVISION AF TEKSTEN -
Maj 2015