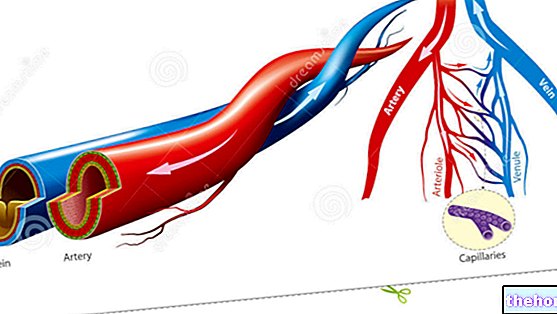
Aktive ingredienser: Brivudine
Brivirac 125 mg tabletter
Hvorfor bruges Brivirac? Hvad er det for?
Brivirac indeholder det aktive stof brivudin. Brivirac har en antiviral virkning og forhindrer den virus, der forårsager St. Anthony's Fire (varicella-zoster-virus) i at formere sig.
Brivirac bruges til voksne, der ikke har abnormiteter i immunsystemet (kroppens forsvar) til tidlig behandling af St. Anthony's Fire (herpes zoster).
Kontraindikationer Når Brivirac ikke bør bruges
Tag ikke Brivirac
- hvis du er allergisk (overfølsom) over for det aktive stof brivudin
- hvis du er allergisk (overfølsom) over for et af de øvrige indholdsstoffer i Brivirac (se afsnit 6)
- hvis du er gravid eller ammer
- hvis du er under 18 år.
Specielt må du IKKE tage Brivirac:
- hvis du tager medicin mod kræft (kemoterapi), især hvis du behandles med:
- 5-fluorouracil (også kaldet 5-FU, et aktivt stof tilhørende gruppen kaldet 5-fluoropyrimidiner)
- cremer, salver, øjendråber eller anden form for medicin til ekstern brug indeholdende 5-fluorouracil
- aktive ingredienser, der omdannes af kroppen til 5-fluorouracil, såsom:
- capecitabin
- floxuridin
- tegafur
- enhver anden aktiv ingrediens i 5-fluoropyrimidin-gruppen
- sammenslutninger af de førnævnte aktive ingredienser
- hvis dit immunsystem (dvs. din krops forsvar mod infektioner) er alvorligt kompromitteret f.eks. hvis du behandles med:
- medicin mod kræft (kemoterapi) eller
- immunsuppressive lægemidler (dvs. medicin, der undertrykker eller nedsætter dit immunsystems funktion)
- hvis du tager en medicin, der indeholder flucytosin til behandling af en svampeinfektion.
- hvis du tager en vorte medicin, der indeholder et aktivt stof fra 5-fluoropyrimidin gruppen.
Forholdsregler ved brug Det, du skal vide, før du tager Brivirac
Tal med din læge eller apotek, før du tager Brivirac.
Tag ikke Brivirac sammen med medicin, der indeholder 5-FU eller andre 5-fluoropyrimidiner (se afsnittene "Tag ikke Brivirac" og "Andre lægemidler og Brivirac").
Tag ikke Brivirac, hvis udslæt allerede er fuldt udviklet (skorpernes begyndelse). Spørg din læge, hvis du er i tvivl.
Spørg din læge til råds, før du tager Brivirac, hvis du har kronisk leversygdom (f.eks. Kronisk hepatitis).
Du bør ikke tage Brivirac i mere end 7 dage, da forlængelse af behandlingsvarigheden ud over den anbefalede tid på 7 dage øger risikoen for at udvikle hepatitis (se også afsnit 4).
Børn og unge
Giv ikke Brivirac til børn og unge mellem 0 og 18 år, da sikkerhed og effekt ikke er undersøgt i denne aldersgruppe.
Interaktioner Hvilke lægemidler eller fødevarer kan ændre effekten af Brivirac
Fortæl det til din læge eller apotek, hvis du tager anden medicin eller har brugt det for nylig. Dette gælder også medicin, som ikke er købt på recept.
BEMÆRK VENLIGST:
Særlig advarsel til patienter behandlet med produkter, der indeholder 5-fluorouracil eller andre 5-fluoropyrimidiner (se også rød boks ovenfor):
Brivirac bør ikke bruges samtidigt med kemoterapimedicin, der indeholder et af følgende aktive stoffer, da de skadelige virkninger af disse lægemidler kan øges kraftigt og være dødelige:
- 5-fluorouracil, herunder former, der skal bruges topisk
- capecitabin
- floxuridin
- tegafur
- andre 5-fluoropyrimidiner
- kombinationer af et hvilket som helst af de ovennævnte stoffer med andre aktive ingredienser.
Tag ikke Brivirac sammen med medicin, der indeholder det aktive stof flucytosin, der bruges til behandling af svampeinfektioner. Tag ikke Brivirac, og kontakt straks din læge, hvis:
- er i behandling baseret på en af ovenstående lægemidler
- du vil blive behandlet med en af ovenstående lægemidler inden for 4 uger efter afslutningen af behandlingen med Brivirac.
Hvis du ved et uheld har taget Brivirac samtidig med en af de ovennævnte lægemidler:
- stoppe med at tage begge lægemidler
- konsultere en læge med det samme. Det kan være nødvendigt at gå til hospitalet for behandling.
Symptomer og tegn på 5-fluorouracil toksicitet på grund af ovenstående interaktioner omfatter:
- utilpashed diarré; betændelse i munden og / eller den indre slimhinde i munden; nedsat antal hvide blodlegemer og knoglemarvsdepression; udslæt og rødme i hele kroppen, med smertefuld hud at røre ved, efterfulgt af store blærer, der fører til store områder af hudafskalning (toksisk epidermal nekrolyse) (se også afsnit 4).
- Erfaring efter markedsføring indikerer en mulig interaktion mellem brivudin og dopaminerge lægemidler mod Parkinsons sygdom, hvilket kan favorisere begyndelsen af et choreaanfald (unormale, ufrivillige, danselignende bevægelser, især arme, ben og ansigt). Brug af Brivirac sammen med mad og drikke Du kan tage Brivirac med eller uden mad.
Advarsler Det er vigtigt at vide, at:
Graviditet og amning
Spørg din læge eller apotek til råds, før du tager medicin.
Brug ikke Brivirac under graviditet.
Brug ikke Brivirac, hvis du ammer. Det aktive stof i Brivirac kan passere til din baby gennem modermælk.
Kørsel og brug af maskiner
Svimmelhed og somnolens er blevet observeret hos nogle patienter, der tager Brivirac, selvom det er usædvanligt. Hvis du bemærker disse bivirkninger, må du ikke køre bil, ikke bruge maskiner eller arbejde uden sikker støtte. Spørg din læge til råds.
Brivirac indeholder lactose
Dette lægemiddel indeholder lactose. Hvis din læge har fortalt dig, at du ikke tåler nogle sukkerarter, skal du kontakte din læge, inden du tager dette lægemiddel.
Dosis, metode og administrationstidspunkt Sådan bruges Brivirac: Dosering
Tag altid denne medicin, som din læge har fortalt dig. Kontakt din læge eller apotek, hvis du er i tvivl.
Den anbefalede dosis er:
1 Brivirac 125 mg tablet en gang dagligt i 7 dage.
Tag Brivirac -tabletten på omtrent samme tidspunkt hver dag.
Brivirac kan tages med eller uden mad.
Synk tabletten hel med en tilstrækkelig mængde væske, f.eks. et glas vand.
Du bør starte behandlingen så hurtigt som muligt, hvilket betyder, at du om muligt skal begynde at tage Brivirac:
- inden for 3 dage efter udseendet af de første hudtegn på St. Anthony's Fire (udslæt) eller
- inden for 2 dage efter udseendet af de første blærer.
Gennemfør den 7 -dages behandling, selvom du har det bedre før.
Hvis symptomerne vedvarer eller forværres i løbet af behandlingsugen, skal du kontakte din læge.
At tage den sædvanlige dosis Brivirac reducerer risikoen for at udvikle postherpetisk neuralgi hos patienter over 50 år. Postpetic neuralgi er vedvarende smerter, der udvikler sig i det område, der tidligere var påvirket af helvedesild, efter at udslæt er blevet bedre.
Behandlingens varighed
Denne medicin er beregnet til kortvarig brug. Det bør kun tages i 7 dage. Tag ikke denne medicin til et andet behandlingsforløb.
Anvendelse til børn og unge
Tag ikke Brivirac, hvis du er under 18 år.
Hvis du har glemt at tage Brivirac
Hvis du har glemt at tage din tablet på dit sædvanlige tidspunkt, skal du tage den, så snart du husker det. Tag den næste tablet den næste dag på omtrent samme tidspunkt som dagen før. Fortsæt med den nye dosis indtil slutningen af 7-kurset behandlingscyklus. dage.
Tag ikke en dobbeltdosis som erstatning for en glemt tablet.
Hvis du gentagne gange glemmer at tage din daglige dosis, skal du fortælle det til din læge.
Hvis du holder op med at tage Brivirac
Stop ikke med at tage Brivirac uden først at konsultere din læge. For at behandlingen skal være fuldt ud effektiv, skal medicinen tages i 7 dage. Spørg din læge eller apoteket, hvis du har yderligere spørgsmål om brugen af denne medicin.
Overdosering Hvad skal jeg gøre, hvis du har taget for meget Brivirac
Hvis du har taget flere tabletter, end du burde, skal du kontakte din læge. Han vil beslutte, om der er behov for yderligere foranstaltninger.
Bivirkninger Hvad er bivirkningerne af Brivirac
Ligesom al anden medicin kan denne medicin forårsage bivirkninger, men ikke alle får bivirkninger.
Stop med at tage Brivirac og fortæl det straks til din læge, hvis du har en allergisk reaktion med tegn og symptomer, herunder kløende eller rød hud (udslæt), øget svedtendens, hævelse (i hænder, fødder, ansigt, tunge, læber, øjenlåg eller strubehoved), besvær ved vejrtrækning (se også afsnit 4) Disse symptomer kan være alvorlige og kræve akut lægehjælp.
Følgende bivirkning er almindeligt observeret (kan forekomme hos op til 1 ud af 10 patienter):
- kvalme (utilpashed).
Følgende bivirkninger er blevet observeret ualmindeligt (kan forekomme hos op til 1 ud af 100 patienter):
- et fald i antallet af en type hvide blodlegemer (granulocytter)
- en stigning i antallet af visse typer hvide blodlegemer (eosinofiler, lymfocytter, monocytter)
- et fald i antallet af røde blodlegemer (anæmi)
- allergiske reaktioner, herunder:
- kløende hud (kløe)
- rødme i huden (erytematøst udslæt)
- øget svedtendens
- hævelse af: hænder, fødder, ansigt, tunge, læber, øjenlåg, strubehoved (larynx ødem)
- hoste, vejrtrækningsbesvær og / eller åndenød
- mangel på appetit
- angst
- søvnløshed, søvnighed
- hovedpine
- svimmelhed
- svimmelhed
- unormale fornemmelser såsom brændende, følelsesløshed, prikkende fornemmelse, oftest i arme og ben (paræstesi)
- forhøjet blodtryk
- fordøjelsesbesvær (dyspepsi), opkastning, mavesmerter
- diarré
- overskydende gas i maven eller tarmen (flatulens)
- forstoppelse
- kronisk leversygdom med ophobning af fedt (fedtlever)
- stigning i blodniveauer af visse stoffer produceret af leveren (stigning i leverenzymer)
- svaghed, træthed (træthed)
- influenzalignende symptomer (utilpashed, feber, smerter og kulderystelser)
Følgende bivirkninger er sjældent set (kan forekomme hos op til 1 ud af 1000 patienter):
- lavt blodtryk
- fald i antallet af blodplader i blodet
- hallucinationer, delirium
- forvirringstilstand
- rysten
- ændret smagssans
- øre smerter
- leverbetændelse (hepatitis), forhøjet bilirubin i blodet
- knoglesmerter
Følgende bivirkninger er også blevet rapporteret, selvom deres hyppighed ikke er kendt (hyppigheden kan ikke estimeres ud fra de tilgængelige data):
- tab af balance
- betændelse i blodkar (vaskulitis)
- hurtigt begyndende leversvigt
- lokaliseret hudbetændelse, der forekommer samme sted i et bestemt tidsrum (fast udslæt), hudbetændelse med eksfoliering (eksfoliativ dermatitis), alvorligt udslæt over hele overfladen af kroppen og inde i munden på grund af allergisk reaktion (erythema multiforme) , sårdannelse i hud, mund, øjne og kønsområder (Stevens Johnsons syndrom).
Indberetning af bivirkninger
Tal med din læge eller apotek, hvis du får bivirkninger, herunder mulige bivirkninger, der ikke er anført i denne indlægsseddel. Du kan også indberette bivirkninger direkte via det nationale rapporteringssystem på: http://www.agenziafarmaco.gov.it/it/responsabili. Ved at rapportere bivirkninger kan du også hjælpe med at give mere information om sikkerheden af dette lægemiddel.
Udløb og opbevaring
Opbevares utilgængeligt for børn.
Brug ikke dette lægemiddel efter den udløbsdato, der står på kartonen og blisterpakningen. Udløbsdatoen refererer til den sidste dag i den pågældende måned.
Opbevar blisterpakningen i den ydre karton for at beskytte medicinen mod lys.
Smid ikke medicin via spildevand eller husholdningsaffald. Spørg din apotek om, hvordan du skal smide medicin, du ikke længere bruger. Dette vil hjælpe med at beskytte miljøet.
Deadline "> Andre oplysninger
Brivirac indeholder
Den aktive ingrediens er brivudin.
1 Brivirac -tablet indeholder 125 mg brivudin.
Øvrige indholdsstoffer er:
- mikrokrystallinsk cellulose
- lactosemonohydrat
- povidon K 24-27
- magnesiumstearat
Hvordan Brivirac ser ud og pakningens indhold
Brivirac 125 mg tabletter er runde, flade, hvide eller næsten hvide med skrå kanter.
Tabletterne findes i en blisterpakning inde i en æske.
Brivirac fås i pakninger med 1 og 7 tabletter og i multipakninger inklusive 5 kartoner, der hver indeholder 7 tabletter.
Ikke alle pakningsstørrelser er nødvendigvis markedsført.
Indlægsseddel: AIFA (Italian Medicines Agency). Indhold offentliggjort i januar 2016. De foreliggende oplysninger er muligvis ikke opdaterede.
For at få adgang til den mest opdaterede version er det tilrådeligt at få adgang til webstedet AIFA (Italian Medicines Agency). Ansvarsfraskrivelse og nyttige oplysninger.
01.0 LÆGEMIDLETS NAVN -
BRIVIRAC 125 MG TABLETTER
02.0 KVALITATIV OG KVANTITATIV SAMMENSÆTNING -
1 tablet indeholder 125 mg brivudin.
Hjælpestof med kendt effekt: lactosemonohydrat. Hver tablet indeholder 37 mg lactosemonohydrat.
Den fulde liste over hjælpestoffer findes i afsnit 6.1
03.0 LÆGEMIDDELFORM -
Tablet
Hvide eller næsten hvide flade tabletter med skrå kanter.
04.0 KLINISKE OPLYSNINGER -
04.1 Terapeutiske indikationer -
Tidlig behandling af akutte herpes zoster -infektioner hos immunkompetente voksne.
04.2 Dosering og indgivelsesmåde -
Dosering
Voksne: en Brivirac -tablet en gang dagligt i syv dage.
Behandlingen bør begynde så hurtigt som muligt, helst inden for 72 timer efter de første hudmanifestationers begyndelse (normalt et "udslæt") eller 48 timer efter den første blisters begyndelse. Tabletterne skal tages på omtrent samme tidspunkt hver dag. Hvis symptomerne vedvarer eller forværres i løbet af de syv dages behandling, skal patienten rådes til at søge lægehjælp. Produktet er beregnet til kortvarig brug.
Denne behandling reducerer også risikoen for at udvikle postherpetisk neuralgi hos patienter over 50 år ved den normale dosis angivet ovenfor (1 tablet Brivirac én gang dagligt i 7 dage).
Efter et første behandlingsforløb (7 dage) bør et andet kursus ikke udføres.
Særlige populationer
Ældre patienter
Ingen dosisjustering er nødvendig hos patienter over 65 år.
Patienter med nedsat nyre- eller leverfunktion
Som en konsekvens af nyre- eller leverinsufficiens observeres ingen signifikante ændringer i systemisk eksponering for brivudin; derfor er dosisjustering ikke nødvendig hos patienter med moderat til svært nedsat nyrefunktion og hos patienter med moderat til svært nedsat leverfunktion (se også afsnit 5.2).
Pædiatrisk population
Brivirac er kontraindiceret til børn i alderen 0 til 18 år, da sikkerhed og effekt i denne aldersgruppe ikke er fastslået (se pkt. 4.3).
Indgivelsesmåde
Oral brug.
Fødeindtag påvirker ikke signifikant absorptionen af brivudin (se pkt. 5.2).
04.3 Kontraindikationer -
Brivirac må ikke administreres i tilfælde af overfølsomhed over for det aktive stof eller over for et eller flere af hjælpestofferne anført i pkt.6.1.
Patienter, der gennemgår antineoplastisk kemoterapi
Brugen af Brivirac er kontraindiceret til patienter, der gennemgår kræftkemoterapi, især hvis de behandles med 5-fluorouracil (5 FU), herunder dets topiske præparater, dets pro-lægemidler (f.eks. Capecitabin, floxuridin, tegafur) og kombinationer, der indeholder disse aktive stoffer, eller andre 5-fluoropyrimidiner (se også pkt. 4.4 og 4.5).
Patienter i svampedræbende behandling med flucytosin
Brugen af Brivirac er kontraindiceret til patienter, der gennemgår antisvampeterapi med flucytosin, da det er et pro-lægemiddel af 5-fluorouracil (5 FU).
Immunkompromitterede patienter
Brugen af Brivirac er kontraindiceret til patienter med immunkompromitteret behandling, f.eks. Patienter, der gennemgår antineoplastisk kemoterapi, immunsuppressiv behandling.
Børn
Sikkerhed og effekt af Brivirac til børn er ikke fastslået, derfor er dets anvendelse ikke indiceret.
Graviditet og amning
Brivirac er kontraindiceret under graviditet eller amning (se også afsnit 4.6).
04.4 Særlige advarsler og passende forholdsregler ved brug -
Brivirac og 5-fluorouracil, herunder dets topiske præparater eller dets lægemidler (f.eks. Capecitabin, floxuridin, tegafur) eller kombinationer indeholdende disse aktive stoffer og andre 5-fluoropyrimidiner (f.eks. Flucytosin) bør ikke administreres samtidigt og et minimumsinterval på 4 uger skal observeres, før behandling påbegyndes med 5-fluoropyrimidin-lægemidler. Som en ekstra sikkerhedsforanstaltning bør DPD-enzymets aktivitet overvåges, før der påbegyndes behandling med 5-fluoropyrimidin-lægemidler hos patienter, der for nylig har fået Brivirac (se også afsnit 4.5 og 4.8).
Brivirac bør ikke anvendes, hvis hudens manifestationer allerede er fuldt udviklet.
Brivirac bør bruges med forsigtighed til patienter med kronisk leversygdom, såsom hepatitis. Data efter markedsføring indikerer, at forlængelse af behandlingen ud over den anbefalede varighed på 7 dage øger risikoen for at udvikle hepatitis (se også pkt.4.8).
Da lactose er til stede blandt hjælpestofferne, bør patienter med sjældne arvelige problemer med galactoseintolerance, Lapp-lactasemangel eller glucose-galactosemalabsorption ikke tage stoffet.
04.5 Interaktioner med andre lægemidler og andre former for interaktion -
Kontraindikationer for samtidig brug af 5-fluorouracil (herunder dets topiske præparater og lægemidler, f.eks. Capecitabin, floxuridin, tegafur) eller andre 5-fluoropyrimidiner såsom flucytosin (se også afsnit 4.3).
Denne interaktion, der resulterer i øget fluoropyrimidintoksicitet, er potentielt dødelig.
Brivudin udøver gennem sin hovedmetabolit bromovinyluracil (BVU) en "irreversibel hæmning af dihydroxypyrimidindehydrogenase (DPD), et enzym, der regulerer metabolismen af både naturlige nukleosider (f.eks. Thymidin) og pyrimidinbaserede lægemidler såsom 5-fluorouracil (5 -FU): Som en konsekvens af hæmning af enzymet er der en overeksponering og øget toksicitet af 5-FU.
Kliniske undersøgelser har vist, at hos raske voksne, der gennemgår et kursus af Brivirac-baseret behandling (125 mg én gang dagligt i 7 dage), sker fuldstændig funktionel genopretning af DPD-enzymaktiviteten 18 dage efter den sidste administration.
Brivirac og 5-fluorouracil eller andre 5-fluoropyrimidiner, såsom capecitabin, floxuridin og tegafur (eller kombinationer, der indeholder disse aktive stoffer) eller flucytosin bør ikke administreres samtidigt, og der bør observeres et minimumsinterval på 4 uger, før lægemiddelbehandling påbegyndes. 5-fluoropyrimidin. Som en ekstra sikkerhedsforanstaltning bør DPD-enzymets aktivitet overvåges, inden der påbegyndes behandling med 5-fluoropyrimidin-lægemidler hos patienter, der for nylig har fået Brivirac.
I tilfælde af utilsigtet administration af 5-FU eller beslægtede lægemidler til patienter behandlet med Brivirac, bør begge lægemidler afbrydes, og drastiske foranstaltninger til reduktion af 5-FU-toksicitet bør implementeres. Umiddelbar hospitalsindlæggelse anbefales, og alle foranstaltninger skal træffes for at forhindre systemiske infektioner og dehydrering. Tegn på 5-FU toksicitet omfatter kvalme, opkastning, diarré og i alvorlige tilfælde stomatitis, mucositis, toksisk epidermal nekrolyse, neutropeni og knoglemarvsdepression.
Dopaminerge lægemidler og / eller Parkinsons sygdom
Erfaring efter markedsføring indikerer en mulig interaktion mellem brivudin og dopaminerge lægemidler mod Parkinsons sygdom, såsom at udfælde chorea.
Andre oplysninger
Der er ikke påvist noget induktions- eller inhiberingspotentiale i det hepatiske P450 -enzymsystem.
Fødeindtag ændrer ikke signifikant absorptionen af brivudin.
04.6 Graviditet og amning -
Brivirac er kontraindiceret under graviditet eller ammende kvinder.
Dyrestudier har ikke vist embryotoksiske eller teratogene virkninger. Giftige virkninger på fosteret blev kun observeret ved høje doser. Sikkerheden ved Brivirac hos gravide er imidlertid ikke fastslået.
Dyreforsøg har vist, at brivudin og dets største metabolit bromovinyluracil (BVU) udskilles i mælk.
04.7 Virkninger på evnen til at føre motorkøretøj og betjene maskiner -
Der er ingen undersøgelser af effekten af Brivirac på evnen til at føre motorkøretøj eller betjene maskiner. Når du kører biler, betjener maskiner eller arbejder uden sikker fodfæste, skal det tages i betragtning, at der i nogle tilfælde er rapporteret om svimmelhed og døsighed (se pkt. 4.8).
04.8 Bivirkninger -
Resumé af sikkerhedsprofilen
Brivudin er blevet administreret til mere end 3900 patienter i kliniske forsøg. Den mest alvorlige, men sjældne, reaktion var hepatitis. Denne reaktion er også blevet observeret under overvågning efter markedsføring.
Den eneste almindelige bivirkning var kvalme (2,1%). De andre hyppigste (usædvanlige og sjældne) bivirkninger var dem relateret til nervesystemet og psykiatriske lidelser SOC. En effekt af brivudin på CNS blev også påvist ved overvågningsdata efter markedsføring .
Hud- og subkutant vævsforstyrrelser er blevet observeret under klinisk brug af produktet, også fremhævet af post-marketing overvågningsdata.
Forekomsten og typen af uønskede reaktioner var sammenlignelige med dem, der vides at forekomme med andre antivirale nukleosidmidler, der tilhører samme klasse.
Oversigtstabel over bivirkninger
Tabellen nedenfor viser bivirkningerne af brivudin grupperet efter system efter faldende sværhedsgrad.
Beskrivelse af udvalgte bivirkninger
Brivudin kan interagere med kemoterapeutiske midler af 5-fluoropyrimidinklassen. Denne interaktion, der inducerer øget fluoropyrimidintoksicitet, er potentielt dødelig (se også 4.4 og 4.5).
Tegn på 5-FU toksicitet omfatter kvalme, opkastning, diarré og i alvorlige tilfælde stomatitis, mucositis, toksisk epidermal nekrolyse, neutropeni og knoglemarvsdepression (se også pkt. 4.5).
Hepatotoksiske virkninger er forekommet både i kliniske forsøg og efter markedsføring. Disse effekter består af kolestatisk eller cytolytisk hepatitis, kolestatisk gulsot eller forhøjede leverenzymer. De fleste tilfælde af hepatitis startede i alderen 3 til 28 dage efter 7-dages afslutning Efter markedsføring viser data, at forlængelse af behandlingen ud over den anbefalede 7-dages periode øger risikoen for hepatitis.
Pædiatrisk population
Brivudin er ikke undersøgt i den pædiatriske population, og dets anvendelse til børn er ikke indiceret. Derfor er sikkerhedsprofilen i den pædiatriske population ukendt.
Indberetning af formodede bivirkninger
Rapportering af formodede bivirkninger, der opstår efter godkendelse af lægemidlet, er vigtig, da det muliggør løbende overvågning af lægemidlets fordel / risiko -balance. Sundhedspersonale anmodes om at rapportere alle formodede bivirkninger via det nationale rapporteringssystem. "Adresse www. agenziafarmaco.gov.it/it/responsabili.
04.9 Overdosering -
Akut overdosis med Brivirac er ikke blevet rapporteret hidtil. Efter forsætlig eller utilsigtet overdosering bør passende symptomatisk og understøttende behandling indledes.
05.0 FARMAKOLOGISKE EGENSKABER -
05.1 "Farmakodynamiske egenskaber -
Antiviral
ATC -kode J05AB15
Brivudin, det aktive stof i Brivirac, er en af de stærkeste nukleosidanaloger, der hæmmer replikationen af Varicella Zoster -virus (VZV). Særligt følsomme er de kliniske stammer af VZV. I virusinficerede celler gennemgår brivudin en række successive phosphoryleringer, der producerer brivudintrifosfat, som er ansvarlig for at hæmme viral replikation.Den intracellulære omdannelse af brivudin til dets phosphorylerede derivater katalyseres af viruskodede enzymer, hovedsageligt thymidinkinase. Phosphorylering forekommer kun. i inficerede celler, hvilket forklarer den høje selektivitet af brivudin til virale mål. Brivudintrifosfat, når det først er dannet i virusinficerede celler, forbliver inde i cellerne i mere end 10 timer og interagerer med viral DNA-polymerase. Denne interaktion resulterer i kraftig hæmning af viral replikation. Resistensmekanismen er baseret. På viral thymidinkinase (TK) I klinisk praksis er kravene til resistens imidlertid kronisk antiviral behandling og patientimmunodefekt, som begge usandsynligt vil forekomme med de angivne indikationer og dosering.
Koncentrationen af brivudin, der er i stand til at hæmme viral replikation in vitro (IC50) svarer til 0,001 mcg / ml (område 0,0003 - 0,003 mcg / ml). Således er brivudin cirka 200 til 1000 gange mere potent end aciclovir og penciclovir til inhibering af VZV -replikation in vitro. Den maksimale plasmakoncentration (Cssmax) for brivudin hos personer, der modtog den foreslåede dosis (125 mg én gang dagligt) er 1,7 mcg / ml (dvs. 1000 gange IC50 "in vitro") og minimumskoncentrationen (Cssmin) er 0,06 mcg / ml (dvs. mindst 60 gange IC50). Brivudin startede meget hurtigt under betingelser med høj viral vækst og nåede 50 % hæmning af viral replikation inden for 1 time efter lægemiddeleksponering. Brivudin udviser også antiviral aktivitet hos forsøgsdyr inficeret med Simian -virus (aber) eller herpes simplex -virus type I (mus og marsvin). Brivudin er aktiv mod herpes simplex -virus type I, mens den ikke har nogen signifikant antiviral aktivitet mod herpes simplex type II. .
Inhibering af virusreplikation understreger effektiviteten af Brivirac til at fremskynde opløsningen af hudmanifestationer hos patienter med den indledende fase af herpes zoster.Den høje in vitro antivirale styrke af brivudin afspejles i den overlegne kliniske effekt, der er observeret i sammenlignende kliniske undersøgelser med aciclovir, vedr. tidsperioden fra behandlingsstart til det sidste vesikulære udbrud: Middeltiden blev reduceret med 25% med brivudin (13,5 timer) sammenlignet med aciclovir (18 timer).
Derudover var den relative risiko for at udvikle postherpetisk neuralgi (PHN) hos immunkompetente patienter over 50 år, der blev behandlet for herpes zoster med brivudin, 25% lavere (33% af patienterne rapporterede PHN) sammenlignet med aciclovir (43% af patienterne rapporterede PHN).
05.2 "Farmakokinetiske egenskaber -
Absorption
Brivudin absorberes hurtigt efter oral administration af Brivirac. Biotilgængeligheden af brivudin er cirka 30% af den orale dosis Brivirac på grund af et forhøjet first -pass metabolisme. Den gennemsnitlige maksimal steady -state plasmakoncentration af brivudin efter administration af en oral dosis på 125 mg Brivirac er 1,7 μg / ml og nås 1 time efter dosis. Fødeindtagelse forsinker lidt absorptionen af brivudin, men påvirker ikke den samlede mængde medicin, der absorberes.
Fordeling
Brivudin fordeler sig i vid udstrækning i væv, hvilket indikeres af det høje distributionsvolumen (75 l). Brivudin er stærkt bundet til plasmaproteiner (> 95%).
Biotransformation
Brivudin metaboliseres i stor udstrækning og hurtigt af enzymet pyrimidinphosphorylase, der spalter kulhydratet for at give bromovinyluracil (BVU), en metabolit uden virustatisk aktivitet. BVU er den eneste metabolit, der påvises i humant plasma og dets maksimale koncentration. Plasma er en faktor på to højere end modersammensætningen.
BVU metaboliseres yderligere til uracyleddikesyre, den største polære metabolit påvist i human urin, men ikke påviselig i plasma.
Eliminering
Brivudin elimineres effektivt med en total kropsclearance på 240 ml / min. Den endelige plasmahalveringstid for brivudin er ca. 16 h. Brivudin udskilles i urinen (65% af den administrerede dosis) primært som uracyleddikesyre og flere polære urinstoflignende forbindelser. Uændret brivudin tegner sig for mindre end 1% af dosis . af Brivirac udskilt i urinen. De kinetiske parametre for BVU, hvad angår terminal halveringstid og clearance, er af samme størrelsesorden som moderforbindelsen.
Linearitet / ikke-linearitet
Lineær kinetik blev observeret over dosisområdet 31,25 til 125 mg.
Steady tilstand for brivudin nås efter 5 dages daglig administration af Brivirac, uden indikation af yderligere efterfølgende akkumulering.
Ældre patienter og patienter med nedsat nyre- eller leverfunktion
De vigtigste kinetiske parametre (AUC, Cmax og terminal plasma-halveringstid) for brivudin målt hos ældre patienter og hos patienter med moderat til svært nedsat nyrefunktion (kreatininclearance mellem 26 og 50 ml / min / 1,73 m2 kropsoverfladeareal og kreatininclearance
05.3 Prækliniske sikkerhedsdata -
Ikke-kliniske data afslører ingen særlig risiko for mennesker ved kortvarig brug baseret på konventionelle undersøgelser af sikkerhedsfarmakologi, genotoksicitet, kræftfremkaldende potentiale, reproduktionstoksicitet.
Prækliniske virkninger af akut og kronisk toksicitet blev observeret i korttidsundersøgelser ved eksponeringer, der blev vurderet tilstrækkeligt over den maksimale humane eksponering.Data indsamlet fra langvarige dyreforsøg med daglig lægemiddeleksponering tæt på det kliniske område, blev ikke anset for signifikante for kortvarig behandling hos mennesker. Målorganet for toksicitet hos alle arter, der blev brugt til prækliniske undersøgelser, var leveren.
06.0 LÆGEMIDDELOPLYSNINGER -
06.1 Hjælpestoffer -
Mikrokrystallinsk cellulose, lactosemonohydrat, povidon K 24-27, magnesiumstearat.
06.2 Uforenelighed "-
Ikke relevant.
06.3 Gyldighedsperiode "-
3 år.
06.4 Særlige opbevaringsforhold -
Opbevar blisterpakningen i den ydre karton for at beskytte den mod lys.
06.5 Den umiddelbare emballages art og emballagens indhold -
a) Beholderens art
Stiv uigennemsigtig PVC -folieblister og aluminiumsfolie.
b) Beholderens indhold
Original pakke med 7 tabletter.
Hospitalspakke med 35 (5 x 7) tabletter.
Prøvepakke med 1 tablet.
Ikke alle pakningsstørrelser er nødvendigvis markedsført.
06.6 Brugsanvisning og håndtering -
Ubrugt medicin og affald fra denne medicin skal bortskaffes i overensstemmelse med lokale regler.
07.0 INDEHAVER AF "MARKEDSFØRINGSTILLADELSEN" -
Laboratori Guidotti S.p.A. - Via Livornese 897, Pisa - La Vettola
Forhandler til salg: A. Menarini Industrie Farmaceutiche Riunite s.r.l. - Via Sette Santi 3, Firenze
08.0 MARKEDSFØRINGSTILLADELSESNUMMER -
AIC n. 035720010 - "125 mg tabletter" 7 tabletter i PVC / AL blister
AIC n. 035720022 - "125 mg tabletter" 35 (5x7) tabletter i PVC / AL blister
09.0 DATO FOR FØRSTE TILLADELSE ELLER FORNYELSE AF TILLADELSEN -
Dato for første godkendelse: 06/07/2000
Dato for den seneste fornyelse: 06/07/2015
10.0 DATO FOR REVISION AF TEKSTEN -
Februar 2016