Definition af thalassæmi
Thalassæmi er en genetisk overført blodsygdom, hvor kroppen syntetiserer en unormal form for hæmoglobin.
Som de fleste ved, er hæmoglobin et protein indeholdt i røde blodlegemer, afgørende for transport af ilt i blodet. Hos personer, der lider af thalassæmi, forårsager den muterede form for hæmoglobin den gradvise, men ubønhørlige ødelæggelse af røde blodlegemer op til anæmi.
Fra de medicinske statistikker er det klart, at thalassæmi hovedsageligt påvirker indbyggerne i mellemøstlige lande, afrikanske lande og alle dem, der befolker sumpede steder (ikke overraskende kaldes thalassæmi også Middelhavsanæmi).
Klassificering og årsager
Ifølge den defekte proteinunderenhed (der udgør hæmoglobin) skelnes der mellem to former for thalassæmi; før vi går videre med analysen, lad os tage et skridt tilbage for at præcisere nogle meget vigtige begreber.
Hæmoglobin er bæreren par excellence, der bruges til at transportere ilt i blodet; det består af to proteiner, kendt som alfa-globulin og beta-globulin.
Thalassæmi opstår, når et eller flere gener, der styrer produktionen af et eller begge disse proteiner, er defekte (muterede).
Thalassæmi er forårsaget af en DNA -mutation af de proteiner, der udgør hæmoglobin: disse ændringer har stor indflydelse på den fysiologiske syntese af hæmoglobin og fører til anæmi ved at ødelægge erytrocytterne.
Klassificeringen af thalassæmi skal foretages på grundlag af to vigtige faktorer:
- Antal muterede gener nedarvet fra forældre
- Type protein involveret (alfa- eller beta -hæmoglobin)
Alpha Thalassæmi
I "alfa" -formen af thalassæmi - hvor de 4 "alfa" kugleformede underenheder af hæmoglobin (ved kromosom 16) kan muteres - er et eller flere defekte gener involveret; hver kugleformede underenhed er klart kodet fra et gen, derfor er gener, der kan være involveret, er 4.

Det generelle symptombillede bliver mere alvorligt, når tre eller fire gener er involveret: i det første tilfælde taler vi om "hæmoglobin H sygdom"(Med moderate eller svære symptomer). Når alle fire gener er involveret, kaldes sygdommen alfa-thalassæmi major: i lignende situationer dør den nyfødte kort før fødslen eller kort tid efter.
Beta Thalassæmi
Beta -formen for thalassæmi opstår, som man kan gætte, når generne, der er involveret i beta -kædernes sammensætning, muteres (på niveau med kromosom 11): i dette tilfælde kan kun to gener påvirkes. Hvis kun et gen ændres, betegnes det som beta-thalassæmi mindre, hvor patienten ikke klager over relevante symptomer. På samme måde som alfa -varianten resulterer involvering af begge gener, der udgør betakæderne af hæmoglobin, i en beta-thalassæmi major (eller Cooley's anæmi), som afspejler alvorlige og alvorlige symptomer; i dette tilfælde begynder symptomerne imidlertid normalt efter et par år efter fødslen.
Se videoen
- Se videoen på youtube
Symptomer
Yderligere oplysninger: Thalassæmi Symptomer
Thalassæmi er en meget alvorlig arvelig sygdom, så meget at nogle af dens varianter, såsom alfa-thalassæmi major, kan få barnet til at dø under fødslen eller kort efter fødslen. Spædbørn med beta-thalassæmi major kan dog overleve og udvikle sig de første symptomer inden for et par år efter fødslen (alvorlig anæmi).
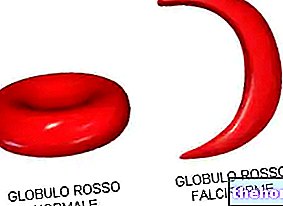
Hvis kun et gen ændres, både i alfa- og beta -formen af thalassæmi, klager patienterne ikke over mærkbare symptomer; kun gennem mikroskopanalysen af en blodprøve taget fra patienten er der en abnormitet i form og struktur på erytrocytterne, meget mindre end normen.
Ud over anæmi kan patienter med thalassæmi opleve et eller flere af følgende symptomer: træthed, humørsvingninger (irritabilitet), vækstsvigt, knogledeformiteter i ansigtet, gulsot, åndenød og mørk urin.
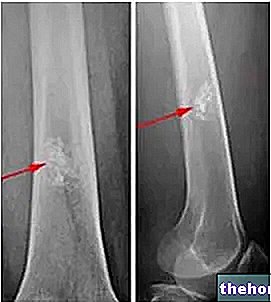
I alvorlige tilfælde kan det symptomatologiske billede af en patient, der lider af thalassæmi, degenerere til det punkt, at der skabes reelle knogledeformiteter, især i ansigtet og kraniet; thalassæmi kan fremme en "unormal ekspansion af knoglemarven, både ved at gøre knoglemassen skrøbelig og ved at øge risikoen for knoglebrud enormt.
Blandt komplikationerne af thalassæmi bør den mulige ophobning af jern (hæmokromatose), et udtryk for både sygdommen selv og for de tilbagevendende blodtransfusioner, som patienten har brug for, også nævnes.
Thalassæmi forårsager ofte splenomegali, det vil sige en overdreven volumetrisk stigning i milten: ofte kræver denne patologiske kliniske tilstand miltomi, kirurgisk fjernelse af organet. Som vi ved, er milten et vigtigt organ, der bruges til syntese af blodlegemer og antistoffer, ud over infektionskontrol: fjernelse, favoriserer klart en nedsættelse af forsvarsfunktionen mod bakterielle og virale fornærmelser, hvilket gør emnet mere følsomt over for infektioner. Det skal dog påpeges, at thalassæmi i sig selv også øger risikoen for at smitte af infektioner: i tilfælde af udskæring af milten i forbindelse med thalassæmi øges chancerne for infektion overdrevet.

Diagnose
Hvis faderen og / eller moderen er ramt af thalassæmi, er sandsynligheden for at overføre sygdommen til afkommet meget stor.Vi har analyseret, at ikke alle former for thalassæmi begynder med en præcis symptomatologi lige fra fødslen: i lignende situationer er det i tilfælde af mistanke om thalassæmi muligt at udsætte patienten for en række specifikke tests og undersøgelser, der er rettet mod den diagnostiske vurdering ( såsom bestemmelse af hæmoglobin A2, som er forhøjet hos raske personer, der bærer beta-thalassæmiske gener).
Blandt de fysiske undersøgelser kan medicinsk palpation af milten undertiden konstatere en thalassæmi: splenomegali udgør som tidligere nævnt et første alarmsignal for middelhavsanæmi. Blodprøver er mere specifikke og præcise: i en blodprøve fra en thalassæmi fremstår de røde blodlegemer, når de ses under et mikroskop, små og har en unormal form. Desuden afslører en omhyggelig blodtælling af en patient, der lider af thalassæmi, alvorlig anæmi: denne test er nyttig til jerntallet i blodet, til at udføre DNA -analyse til diagnostisk vurdering af sygdommen og til at evaluere den mulige mutation af "hæmoglobin" .
På den anden side afslører elektroforese af hæmoglobiner den unormale form af de iltbærende proteiner.
Nogle varianter af thalassæmi kan ikke diagnosticeres med elektroforese: i dette tilfælde vil patienten blive udsat for en "mutationsanalyse" test, som er nyttig til at detektere og fastslå thalassæmi.
Medicin og behandlinger
Se også: Medicin til behandling af thalassæmi
Da det er en genetisk overført sygdom, er det forståeligt, at der - i øjeblikket - ikke er noget stof, der er i stand til at vende sygdommen; dog er det muligt at kontrollere symptomerne og forbedre patientens livskvalitet. Valget af en behandling frem for en anden afhænger af typen af thalassæmi og symptomernes sværhedsgrad.
I den milde variant af thalassæmi (hvor f.eks. Kun et gen ændres) er der ikke behov for medicin, da patienten ikke klager over symptomer. Under sådanne omstændigheder er det stadig tilrådeligt at udføre de nødvendige kontroller regelmæssigt; Lejlighedsvis er lejlighedsvis blodtransfusioner nogle gange nyttige (især ved operation og fødsel).
Ved moderate eller svære symptomatiske former er behandlingsmetoden anderledes og kan kræve hyppige blodtransfusioner eller i alvorlige tilfælde stamcelletransplantation.
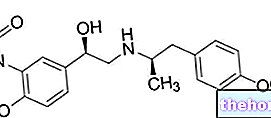
- Blodtransfusioner: denne terapeutiske tilgang kan også skabe alvorlige komplikationer, da hyppige transfusioner kan favorisere en patologisk ophobning af jern i blodet (hæmokromatose), som kræver specifik behandling med det formål at fjerne jernopbevaring, kendt som terapikelator (med lægemidler som Deferasirox og Deferiprone). For yderligere information: læs artiklen om lægemidler til behandling af hæmokromatose.
- Knoglemarvstransplantation: forbeholdt de mest alvorlige tilfælde, hvor thalassæmi skaber alvorlige dysfunktioner i kroppen.