Almindelighed
Osteogenesis imperfecta er en medfødt genetisk sygdom, der ikke er knyttet til køn, ansvarlig for en vis knogleskørhed og en markant tendens til brud.
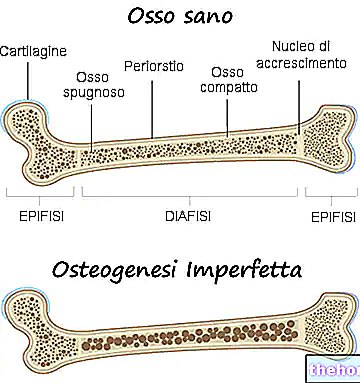
Symptomerne på osteogenesis imperfecta er mange; generelt består de af: knoglesvækkelse, høj tendens til knoglebrud, tilstedeværelse af blå, grå eller lilla okulære sclerae, tilstedeværelse af knogledeformiteter eller andre skeletforandringer, trekantet ansigt, tandskørhed osv. .
Generelt er følgende afgørende for en korrekt diagnose af osteogenesis imperfecta: fysisk undersøgelse, sygehistorie, medicinsk billeddannelsestest, en type I kollagenvurderingstest og en genetisk test.
Desværre er de eneste behandlinger, der er tilgængelige for patienter med osteogenesis imperfecta, i øjeblikket symptomatiske. Den pågældende sygdom er faktisk uhelbredelig.
Hvad er osteogenesis imperfecta?
Osteogenesis imperfecta er en genetisk sygdom, der gør den berørte persons knogler svagere og mere tilbøjelige til brud.
I virkeligheden, med udtrykket osteogenesis imperfecta, refererer læger til en heterogen gruppe af genetiske sygdomme, kendetegnet ved en vis grad af knogleskørhed. Der er derfor flere former (eller typer) af osteogenesis imperfecta, nogle meget mere alvorlige end andre.
DET ER EN KONGENITAL SYGDOM
Hos mennesker, der er ramt af det, er osteogenesis imperfecta en sygdom, der er til stede fra fødslen, og derfor kan den for alt i verden defineres som en medfødt sygdom.
ER DET SEX RELATERET?
Osteogenesis imperfecta er ikke en kønsrelateret genetisk sygdom, såsom hæmofili eller Klinefelters syndrom.
EPIDEMILOGI
Ifølge nogle statistiske undersøgelser ville forekomsten af osteogenesis imperfecta være lig med et tilfælde for hver 15.000-20.000 fødsler. Det betyder, at hver 15.000-20.000 nyfødte får en ramt af osteogenesis imperfecta.
Andre statistiske undersøgelser har også vist, at osteogenesis imperfecta påvirker mænd og kvinder lige meget, og at det ikke foretrækker en bestemt befolkning eller etnisk gruppe.
Levetid er en ekstremt variabel parameter, som afhænger af formen for osteogenesis imperfecta.
Årsager
Osteogenesis imperfecta skyldes næsten altid en kvalitativ og kvantitativ ændring af produktionen af type I -kollagen.
Type I -kollagen er afgørende for at styrke knogler og for at opretholde sunde bindevæv, der udgør brusk, sener, hud, okulær sclera osv.
Derfor påvirker en ændring i produktionen af type I -kollagen styrken af knoglerne og bindevævets gode helbred i menneskekroppen.
HVAD ÆNDRER COLLAGEN -PRODUKTIONEN?
En genetisk sygdom er en tilstand, der opstår på grund af en mutation af et eller flere gener, der udgør cellulært DNA.
I tilfælde af osteogenesis imperfecta findes årsagerne til sidstnævnte næsten altid i mutationen af en eller begge generne COL1A1 (placeret på kromosom 17) og COL1A2 (placeret på kromosom 7).
Under normale forhold regulerer COL1A1 og COL1A2 den normale produktion af type I kollagen; i nærvær af mutationer i deres ladning fejler de i deres regulerende funktion.
Vigtigt: Hvilke andre gener, hvis de muteres, forårsager osteogenesis imperfecta?
Ud over mutationerne af COL1A1 og COL1A2 er mutationer i IFITM5-, SERPINF1-, CRTAP- og LEPRE1 -generne potentielle årsager til osteogenesis imperfecta.
De førnævnte gener dækker funktioner, der er forskellige fra COL1A1 og COL1A2 - derfor styrer de ikke produktionen af type I -kollagen - men de har stadig en "indflydelse på styrken og modstanden i knoglerne i det menneskelige skelet.
HVILKEN GENETISK SYGDOM ER DET?
Osteogenesis imperfecta er en autosomal genetisk sygdom.
Udtrykket autosomalt, forbundet med en genetisk sygdom, indikerer, at den pågældende tilstand skyldes genetiske mutationer baseret på autosomale og ikke-kønskromosomer.
Læserne mindes om, at mennesket besidder et kromosomalt sæt på 23 par samlede kromosomer, hvor 22 par er af autosomal type og kun et par er af den seksuelle type Par af kromosomer af den seksuelle type påvirker køn individ.
Osteogenesis imperfecta efter mutationer i COL1A1, COL1A2 og IFITM5 har alle karakteristika ved en autosomal dominerende sygdom.Når det skyldes mutationer i generne SERPINF1, CRTAP og LEPRE1, har det karakteristika ved en autosomal recessiv sygdom.
TYPER
I øjeblikket mener læger, at der er 8 typer (eller former) for osteogenesis imperfecta. For at skelne de forskellige typer besluttede de at bruge den romerske nummerering, for at være præcis de første otte romertal.
Tabellen nedenfor viser de 8 former for osteogenesis imperfecta, de mutationer, der forårsager dem og andre egenskaber.
Fyr
Muteret gen
Type genetisk sygdom
DET
COL1A1
Autosomal dominerende
II
COL1A1 og COL1A2
Autosomal dominerende
III
COL1A1 og COL1A2
Autosomal dominerende
IV
COL1A1 og COL1A2
Autosomal dominerende
V.
IFITM5
Autosomal dominerende
DU
SERPINF1
Autosomal recessiv
VII
CRTAP
Autosomal recessiv
VIII
HARE 1
Autosomal recessiv
* NB: naturligvis er mutationerne i COL1A1 og COL1A2, der forårsager de første fire former for osteogenesis imperfecta, genetiske ændringer med lidt forskellige egenskaber. Ellers ville det ikke give nogen mening at skelne den ene fra den anden.
Symptomer, tegn og komplikationer
Alle former for osteogenesis imperfecta er ansvarlige for en svækkelse af knoglerne, således at den person, der er ramt af sygdommen, har en særlig disposition for brud. Graden af svækkelse af knoglerne varierer alt efter formen; for nogle af disse er denne svækkelse større end for andre.
Når det er sagt, skal det påpeges, at hver form for osteogenesis imperfecta har sit eget symptomatiske billede, som for nogle måske husker det symptomatologiske billede af andre former.
MULIGE symptomer og tegn
De mulige symptomer og tegn på osteogenesis imperfecta omfatter:
- Tilstedeværelse af knoglemisdannelser;
- Tilstedeværelse af en kort og lille krop (beregnet som bagagerum);
- Ledproblemer (f.eks .: løse led);
- Muskelsvaghed;
- Blå, lilla eller grå okulær sclera;
- Trekantet ansigt;
- Tønde bryst;
- Morfologiske anomalier i rygsøjlen;
- Tandskørhed;
- Nedgang eller totalt tab af hørelse;
- Åndedrætsproblemer
- Problemer relateret til fravær eller mangel på type 1 -kollagen.
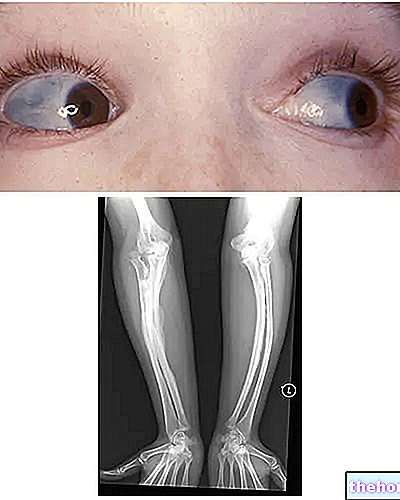
Osteogenesis Imperfecta: bemærk den blå farve af sclerae og de knogledeformationer, der kendetegner sygdommen. Fra wikipedia.org
HVAD ER DE MEST ALVORLIGE FORMER FOR IMPERFEKT OSTEOGENESIS?
Læger klassificerer den symptomatologiske sværhedsgrad af de forskellige typer osteogenesis imperfecta på en skala på 3 grader, som er: den milde grad, den moderate grad og den alvorlige grad.
Kun én form tilhører kategorien "mild grad": "type I osteogenesis imperfecta"; 4 former for osteogenesis imperfecta tilhører kategorien "moderat grad": IV, V og VI; endelig hører kategorien "alvorlig grad" til 3 former: II, III, VII og VIII.
TYPE I: FUNKTIONER
Den mest almindelige og mindst alvorlige form af alle, type I osteogenesis imperfecta har følgende egenskaber:
- Det forårsager brud især før puberteten;
- Det har "næsten ingen indflydelse på højden, så patienterne er normalt af normal" højde;
- Forårsager ledproblemer og muskelsvaghed
- Det er ansvarligt for blå, lilla eller grå sclera;
- Det er årsag til trekantede ansigts- og rygmarvsanomalier;
- Det forårsager næsten aldrig knogledeformiteter. Hvis det provokerer dem, er de minimale;
- Det kan forårsage tandskørhed og / eller høretab (sidstnævnte forekommer normalt i voksenalderen);
- Det er forbundet med tilstedeværelsen af type I kollagen, som er normal i kvalitet, men unormal i mængde (det er dårligere end normalt).
TYPE II: FUNKTIONER
Type II osteogenesis imperfecta er kendetegnet ved:
- Dødsårsag ved fødslen eller kort tid efter. Åndedrætsproblemer forårsager næsten altid død;
- Tilstedeværelse af betydelig knogleskørhed og alvorlige knogledeformiteter;
- Kort statur og underudviklede lunger
- Blå, lilla eller gråfarvet sclera;
- Tilstedeværelse af kvantitative og kvalitative anomalier af type I kollagen.
TYPE III: FUNKTIONER
Type III osteogenesis imperfecta har følgende egenskaber:
- Selvom det er meget alvorligt, forårsager det ikke ofte død i den nyfødte periode;
- Det er forbundet med "høj knogleskørhed;
- Den er ansvarlig for kort statur, ledproblemer, muskelsvaghed (især i ben og arme), tøndebryst, trekantet ansigt og unormal krumning af rygsøjlen;
- Det skyldes blå, lilla eller grå sclera;
- Det kan forårsage vejrtrækningsproblemer, tandskørhed og høretab;
- Den er ofte ansvarlig for knogledeformationer;
- Det er forbundet med kvalitative og kvantitative abnormiteter af type I kollagen.
TYPE IV: FUNKTIONER
Type IV osteogenese er kendetegnet ved:
- En grad af knogleskørhed mellem form II og III og form I;
- Kortere end gennemsnittet;
- Blå, lilla eller gråfarvet sclera;
- Knogledeformiteter af mild / moderat enhed, små abnormiteter i rygsøjlen og tøndebrystet;
- Trekantet ansigt;
- Mulig tilstedeværelse af tandskørhed og høretab;
- Tilstedeværelse af type I kollagen abnormiteter.
TYPE V: FUNKTIONER
Type V osteogenesis imperfecta ligner type IV osteogenesis imperfecta på nogle måder. Det har dog nogle særegenheder, som er:
- Normalfarvet sclera;
- Fravær af dental skrøbelighed;
- Dannelse af unormale knoglede hård hud under helingsprocessen af knækkede knogler;
- Forkalkning af den interosseøse membran, der befinder sig mellem radius og ulna. Dette forringer underarmens mobilitet.
TYPE VI: FUNKTIONER
Også type VI osteogenesis imperfecta ligner form IV. For at skelne den fra sidstnævnte er nogle særegenheder, herunder de høje blodniveauer af alkalisk fosfatase og tilstedeværelsen på nogle knogler af lameller (knoklet) svarende til rygsøjler af fisk.
TYPE VII: FUNKTIONER
Symptomatisk kan type VII osteogenesis imperfecta i visse tilfælde ligne type IV og type II i andre tilfælde.
Det særlige ved denne alvorlige patologiske form omfatter:
- Kort statur;
- Tilstedeværelsen af en ekstremt kort humerus (armben) og lårben (lårben);
- Den hyppige tilstedeværelse af en hofte deformitet, kendt som coxa vara.
TYPE VIII: KARAKTERISTIKA
Type VIII osteogenesis imperfecta minder meget om form II og III.
Blandt sine særlige kendetegn skiller følgende sig ud: det alvorlige vækstunderskud, den alvorlige skelethypomineralisering og fraværet (eller den knappe tilstedeværelse) af prolyl 3-hydroxylase-enzymet.
Diagnose
Generelt begynder den diagnostiske proces, som patienter med en formodet form for osteogenesis imperfecta udsættes for, med en omhyggelig fysisk undersøgelse og en omhyggelig sygehistorie; derefter fortsætter det med en "analyse af patientens familiehistorie og med en række diagnostiske billeddannelsestest (røntgenstråler, CT-scanninger osv.); endelig ender det med en kvantitativ og kvalitativ evaluering af type I-kollagen og med en genetisk test.
I dag er der mulighed for at diagnosticere osteogenesis imperfecta selv i prænatalfasen ved at udsætte en gravid kvinde for ultralyd.
VIGTIGHEDEN FOR MÅL EKSAMINATIONEN OG HISTORIEN
En lægeekspert i osteogenesis imperfecta er i stand til meget ofte at diagnosticere den førnævnte sygdom selv kun ved hjælp af en fysisk undersøgelse og anamnese. Det betyder, at disse diagnostiske tests ikke er ubetydelige.
EVALUERING AF TYPE I COLLAGEN -PRODUKTION
Som regel er den kvalitative og kvantitative vurdering af type I -kollagen en meget pålidelig test, da de fleste tilfælde af osteogenesis imperfecta som nævnt er kendetegnet ved mutationer i de gener, der styrer produktionen af type 1 -kollagen.
For at vurdere mængden og kvaliteten af type I kollagen til stede på mobilniveau hos en person, kan læger stole på en hudbiopsi eller en bestemt blodprøve.
Begge disse evaluerende tests er ret komplekse, og patienten (eller hans forældre) må muligvis vente flere uger for at kende resultaterne.
GENETISK TEST
Gennem en genetisk test, der undersøger hele det individuelle DNA, der undersøges, er lægerne i stand til definitivt at bestemme egenskaberne ved den tilstedeværende genetiske mutation.
Generelt forventes udførelsen af en genetisk test på alt cellulært DNA, når evalueringen af karakteristika for type I -kollagen ikke har givet de ønskede resultater, eller når det ikke er en mutation i COL1A1 eller COL1A2, der forårsager "osteogenesis imperfecta.
PRENATAL DIAGNOSE
Prænatal ultralyd er meget nyttig til at identificere type II og type III osteogenesis imperfecta.
Terapi
Der er i øjeblikket ingen specifik kur mod osteogenesis imperfecta. Med andre ord er mennesker med osteogenesis imperfecta bestemt til at leve med den førnævnte tilstand indtil døden, hvilket ofte skyldes konsekvenserne af selve sygdommen.
Manglen på specifik terapi udelukker ikke eksistensen af andre behandlingsformer. Blandt de terapeutiske muligheder for en patient med osteogenesis imperfecta er forskellige symptomatiske behandlinger faktisk inkluderet; Med symptomatiske terapier mener vi behandlinger, der er i stand til at lindre symptomer, bremse sygdomsforløbet og forhindre (eller i det mindste udsætte) de alvorligste konsekvenser.
MULIGE SYMPTOMATISKE BEHANDLINGER
På listen over mulige symptomatiske behandlinger for osteogenesis imperfecta skiller følgende sig ud:
- Den kirurgiske indsættelse inde i de længste knogler (N.B: den mest tilbøjelige til brud) af negle, der giver større modstand mod brud og deformiteter. Denne operation kaldes rodning intramedullær;
- Konservativ eller kirurgisk behandling af brud og / eller knogledeformiteter;
- Tandpleje, for at beskytte tændernes sundhed;
- Smertelindrende behandlinger i tilfælde af meget smertefulde multiple frakturer;
- Fysioterapi, til forlængelse og styrkelse af muskler Et elastisk og tonisk muskulaturapparat giver dig mulighed for at forhindre fald, der kan føre til forskellige knoglebrud;
- Brug af hjælpemidler til bevægelse, herunder kørestole, seler, krykker osv.
FORDELE VED BEVÆGELSE
For personer med osteogenesis imperfecta anbefaler læger konstant øvelse af fysisk træning og bevægelse generelt, da begge disse aktiviteter bidrager til at styrke skelet- og muskelsystemerne.
Blandt de anbefalede sportsgrene er: svømning, da det er en "lav påvirkning fysisk aktivitet på skeletsystemet" og gåture.
FORDELE FRA EN SUND LIVSTIL
At føre et sundt liv, undgå at ryge, drikke for meget alkohol, spise for meget og dårligt osv., Har mere end diskrete sundhedsmæssige fordele for patienter med osteogenesis imperfecta, da det bremser sygdommens forløb og reducerer knogleskørhed.
SYMPTOMATISKE BEHANDLINGER I EKSPERIMENTERINGSFASEN
I øjeblikket evaluerer læger og forskere effektiviteten af nogle symptomatiske behandlinger, herunder behandling med væksthormon og bisphosphonatbaseret intravenøs og oral behandling.
I øjeblikket lover resultaterne fra de førnævnte undersøgelsesbehandlinger godt for hele det medicinske samfund.
Prognose
Osteogenesis imperfecta er en sygdom med en negativ prognose, da den er uhelbredelig, kompromitterer livskvaliteten drastisk og i nogle tilfælde forårsager for tidlig død af det berørte emne.
Det skal dog bemærkes, at mange mennesker med en mild form for osteogenesis imperfecta også takket være moderne symptomatiske behandlinger er i stand til at leve et behageligt og tilfredsstillende liv.
Forebyggelse
Desværre er der i øjeblikket ingen forebyggende foranstaltning mod osteogenesis imperfecta.