Aktive ingredienser: Erythropoietin (epoetin alfa)
EPREX 2.000 IE / ml, 4.000 IE / ml, 10.000 IE / ml og 40.000 IE / ml INJEKTIONSLØSNING I FØRFyldte sprøjter
Hvorfor bruges Eprex? Hvad er det for?
Hvad er EPREX, og hvad skal det bruges til
EPREX indeholder det aktive stof epoetin alfa, et protein, der stimulerer knoglemarven til at producere flere røde blodlegemer, celler, der bærer hæmoglobin (et stof, der kan transportere ilt). Epoetin alfa er en kopi af humant erythropoietin og virker på den måde.
- EPREX bruges til behandling af symptomatisk anæmi forårsaget af nyresvigt.
- hos børn i hæmodialyse
- hos voksne i hæmodialyse og peritonealdialyse
- hos voksne med svær anæmi, der endnu ikke er under dialyse.
Hvis du har nyresvigt, og dine nyrer ikke producerer nok erythropoietin (hvilket er nødvendigt for produktionen af røde blodlegemer), kan du have få røde blodlegemer i dit blod. EPREX er ordineret til at stimulere knoglemarven til at producere flere røde blodlegemer.
- EPREX bruges til behandling af anæmi, der kan opstå under kemoterapi for solide tumorer, malignt lymfom eller myelomatose (knoglemarvskræft), hvis din læge mener, at du muligvis skal have blodtransfusion. EPREX kan reducere behovet for transfusioner.
- EPREX bruges til at behandle moderat anæmiske patienter, der er kandidater til at deponere deres blod i påvente af operation, så det kan transfunderes under eller efter operationen. Da EPREX stimulerer produktionen af røde blodlegemer, er det muligt at trække blod fra disse mennesker en større mængde blod.
- EPREX bruges til at behandle moderat anæmiske voksne patienter, der gennemgår større ortopædkirurgi (f.eks. Hofte- eller knæudskiftning) for at reducere behovet for blodtransfusioner.
Kontraindikationer Når Eprex ikke bør bruges
Brug ikke EPREX
- Hvis du er allergisk (overfølsom) over for epoetin alfa eller et af de øvrige indholdsstoffer i EPREX (angivet under pakningens indhold og andre oplysninger)
- Hvis du er blevet diagnosticeret med "Pure Red Cell Aplasia (knoglemarven kan ikke lave nok røde blodlegemer) efter tidligere behandling med et stof, der stimulerer produktionen af røde blodlegemer (inklusive EPREX). Se afsnittet Mulige bivirkninger.
- Hvis du har problemer med ukontrolleret forhøjet blodtryk
- At stimulere produktionen af røde blodlegemer (så der kan trækkes mere blod fra dig), hvis du ikke kan modtage transfusioner af dit eget blod under eller efter operationen.
- Hvis du skal foretage valgfri større ortopædkirurgi (f.eks. Hofte- eller knæudskiftning) og:
- har alvorlig hjertesygdom
- du har alvorlige problemer med dine vener og arterier
- for nylig har haft et hjerteanfald eller slagtilfælde
- kan ikke tage blodfortyndende medicin
EPREX er muligvis ikke egnet til dig. Diskuter dette med din læge. Mens du bruger EPREX kan nogle mennesker have brug for medicin for at reducere risikoen for blodpropper.Hvis du ikke kan tage antikoagulant medicin, bør du ikke tage EPREX.
Forholdsregler ved brug Hvad du skal vide, før du tager Eprex
Vær særlig forsigtig med EPREX
EPREX og andre stimulerende midler til røde blodlegemer kan øge risikoen for at udvikle blodpropper hos alle patienter.Denne risiko kan være højere, hvis du har andre risikofaktorer for at udvikle blodpropper (f.eks. Hvis du tidligere har haft en blodprop eller er overvægtig, har diabetes, har hjertesygdomme eller har været immobiliseret i lang tid pga. operation eller sygdom). Fortæl din læge om nogen af disse ting. Din læge hjælper dig med at beslutte, om EPREX er det rigtige for dig.
Det er vigtigt, at du taler med din læge, hvis noget af det følgende gælder for dig.
Du kan stadig bruge EPREX, men du bør først diskutere dette med din læge.
- Hvis du ved, at du har smerter eller har lidt af:
- Højt blodtryk;
- Anfald eller anfald
- Lever sygdom;
- Anæmi fra andre årsager;
- Porfyri (en sjælden blodsygdom).
- Hvis du har kræft, skal du være opmærksom på, at stoffer, der stimulerer produktionen af røde blodlegemer (f.eks. EPREX), kan fungere som en vækstfaktor og teoretisk påvirke tumorprogressionen. En blodtransfusion kan være at foretrække afhængigt af din individuelle tilstand. Diskuter dette med din læge.
Vær særlig opmærksom på stoffer, der stimulerer produktionen af røde blodlegemer:
EPREX tilhører en gruppe stoffer, der stimulerer produktionen af røde blodlegemer som menneskelige erytropoietiske faktorer. Lægen vil altid sørge for at registrere det nøjagtige navn på det produkt, han bruger. Hvis du under din behandling får et stof, der tilhører samme gruppe, men som er forskelligt fra EPREX, bedes du kontakte din læge eller apotek før brug.
Interaktioner Hvilke lægemidler eller fødevarer kan ændre virkningen af Eprex
EPREX forstyrrer normalt ikke andre lægemidler, men fortæl det altid til din læge, hvis du tager eller for nylig har taget anden medicin, også medicin, der ikke kræver recept.
Hvis du tager en medicin kaldet cyclosporin (bruges f.eks. Efter nyretransplantation), kan din læge bestille blodprøver for at kontrollere cyclosporinniveauerne under behandling med EPREX.
Jerntilskud og andre anti-anæmiske faktorer kan øge effektiviteten af EPREX. Din læge vil beslutte, om du skal tage dem.
I tilfælde af hospitalsindlæggelse eller lægeundersøgelse bedes du informere om, at du er i behandling med EPREX. Dette kan påvirke andre behandlinger eller testresultater.
Advarsler Det er vigtigt at vide, at:
Graviditet og amning
Det er vigtigt at fortælle din læge, om en af følgende betingelser gælder for dig. Du kan stadig bruge EPREX, men diskuter dette først med din læge.
- Hvis du er gravid, eller tror du kan være.
- Hvis du ammer.
Dosis, metode og administrationstidspunkt Sådan bruges Eprex: Dosering
Brug altid denne medicin nøjagtigt som anvist af din læge. Spørg din læge, hvis du er i tvivl.
Din læge har på baggrund af dine blodprøver fastslået, at du har brug for EPREX.
EPREX kan gives ved injektion:
- Ind i en vene eller et rør i en vene (intravenøst)
- Under huden (subkutant)
Din læge vil beslutte, hvordan EPREX skal administreres. Injektioner gives normalt af en læge, sygeplejerske eller læge; nogle mennesker kan lære at administrere stoffet subkutant på egen hånd: se Instruktioner til selv at injicere EPREX.
- Eprex bør ikke bruges:
- efter udløbsdatoen angivet på etiketten eller den ydre karton
- hvis du ved eller tror, at medicinen ved et uheld er blevet frosset ned eller ja
- og der var et køleskab fejl
Dosen af EPREX er baseret på din kropsvægt i kilogram og vælges af din læge afhængigt af årsagen til anæmi.
Din læge vil kontrollere dit blodtryk regelmæssigt under behandling med EPREX.
Patienter med nyreinsufficiens
- Din hæmoglobinværdi opretholdes mellem 10 og 12 g / dl, da højere hæmoglobinniveauer kan øge risikoen for trombose og død.
- Den sædvanlige startdosis af EPREX til voksne eller børn er 50 internationale enheder (IE) pr. Kg (/ kg) kropsvægt, givet 3 gange om ugen.
- Hos patienter i peritonealdialyse kan administration foretages to gange om ugen.
- EPREX administreres intravenøst (vene eller rør i en vene) til voksne og børn. Når den intravenøse vej (vene eller slange i en vene) ikke er tilgængelig, kan din læge beslutte, om EPREX skal injiceres under huden (subkutant). Herunder dialysepatienter og patienter, der endnu ikke er i dialyse.
- Din læge vil have regelmæssige blodprøver for at kontrollere, om din anæmi reagerer, og kan justere dosis, normalt ikke oftere end en gang hver 4. uge.
- Når anæmi er korrigeret, vil din læge fortsætte med at kontrollere dine blodprøver regelmæssigt. Dosen af EPREX og hyppigheden af administration kan justeres yderligere for at opretholde responsen på behandlingen.
- Hvis du behandles med EPREX med længere dosisintervaller (større end en gang om ugen), er du muligvis ikke i stand til at opretholde dine hæmoglobinniveauer tilstrækkeligt, og du skal muligvis øge din EPREX -dosis eller administrationens hyppighed.
- For at gøre behandlingen mere effektiv kan jerntilskud være nyttige for dig før og under behandling med EPREX.
- Hvis du er i hæmodialyse, når du starter behandling med EPREX, skal din dialysebehandling muligvis justeres. Lægen bestemmer.
Voksne patienter i kemoterapi
- Din læge kan starte behandling med EPREX, hvis dit hæmoglobinniveau er 10 g / dl eller mindre.
- Din hæmoglobinværdi opretholdes mellem 10 og 12 g / dl, da højere hæmoglobinniveauer kan øge risikoen for trombose og død.
- Startdosis er 150 IE pr. Kg legemsvægt 3 gange om ugen eller 450 IE pr. Kg legemsvægt en gang om ugen.
- EPREX administreres ved subkutan injektion.
- Din læge vil have regelmæssige blodprøver og kan justere dosis afhængigt af dit svar på behandling med EPREX.
- For at gøre behandlingen mere effektiv kan et jerntilskud være nyttigt for dig før og under behandlingen med EPREX.
- Behandlingen med EPREX fortsættes normalt i 1 måned efter afslutningen af kemoterapien.
Voksne patienter, der deponerer deres eget blod
- Den sædvanlige dosis er 600 IE pr. Kg legemsvægt 2 gange om ugen.
- EPREX indgives i en vene umiddelbart efter deponering af blod i 3 uger før operationen.
- For at gøre behandlingen mere effektiv kan et jerntilskud være nyttigt for dig før og under behandlingen med EPREX.
Voksne patienter, der er kandidater til større ortopædkirurgi
- Den anbefalede dosis er 600 IE pr. Kg legemsvægt en gang om ugen.
- EPREX administreres ved subkutan injektion hver uge i 3 uger før operationen og på operationsdagen.
- Hvis det er nødvendigt at reducere tiden før operationen, får du en daglig dosis på 300 IE / kg i de 10 dage forud for operationen, på operationsdagen og i de 4 dage efter operationen.
- Hvis blodprøverne før operationen afslører for høje hæmoglobinværdier, stoppes behandlingen.
- For at gøre behandlingen mere effektiv kan jerntilskud være nyttige for dig før og under behandling med EPREX.
Instruktioner til selv at injicere EPREX
- I starten af behandlingen gives EPREX normalt af en læge eller sygeplejerske, og derefter kan din læge foreslå, at du (eller din plejer) lærer at injicere (subkutant).
- Forsøg ikke at injicere dig selv, hvis din læge eller sygeplejerske ikke har fortalt dig hvordan.
- Følg altid instruktionerne fra din læge eller sygeplejerske.
- Brug kun EPREX, hvis det er blevet opbevaret korrekt - se afsnittet Sådan opbevares EPREX
- Fjern Eprex-sprøjten før brug, og lad den nå stuetemperatur. Det tager normalt 15-30 minutter.
Træk en enkelt dosis EPREX ud af hver fyldt injektionssprøjte. Når EPREX administreres under huden (subkutant), overstiger volumen normalt ikke 1 milliliter (1 ml) for en enkelt injektion.
EPREX skal administreres alene og ikke blandes med andre injektionsvæsker.
Ryst ikke EPREX fyldte sprøjter. Langvarig kraftig rystning kan beskadige produktet. Brug ikke produktet, hvis det er rystet kraftigt.
Sådan injiceres du selv ved hjælp af de fyldte sprøjter
De fyldte sprøjter er udstyret med en nålesikkerhedsanordning, PROTECS ™, for at forhindre risiko for at nålen klistrer efter brug. Dette er angivet på emballagen.
- Fjern den fyldte injektionssprøjte fra køleskabet før brug. Væsken skal nå stuetemperatur. Fjern ikke kanyledækslet, mens du venter på, at det når stuetemperatur.
- Kontroller den fyldte sprøjte for at sikre, at den er den rigtige dosis, at den ikke er udløbet, at den ikke er beskadiget, og at væsken er klar og ikke frosset.
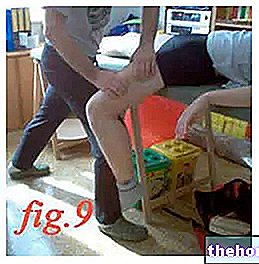
- Vælg injektionsstedet. De mest egnede steder til injektion er øvre lår og mave, undtagen området omkring navlen. Skift injektionssted hver gang.
- At vaske hænder. Brug en antiseptisk klud til at desinficere injektionsstedet.
- Hold den fyldte sprøjte ved sprøjtecylinderen med den dækkede kanyle pegende opad.
- Hold ikke i stempelhovedet, stemplet, nålebeskyttelsesfløjen eller nålebeskyttelseshætten.
- Træk under ingen omstændigheder stemplet tilbage
- Fjern ikke kanylehætten på den fyldte sprøjte, før du er klar til at administrere EPREX
- Fjern beskyttelseshætten fra sprøjtenålen ved at holde den i kroppen og trække i hætten uden at vride den. Skub ikke på stemplet, rør ved nålen eller ryst sprøjten. - Rør ikke ved aktiveringsclipsene til sikkerhedsanordningen for at forhindre for tidligt nåldæksel med nålebeskyttelsesklappen
- Løft huden mellem tommelfinger og pegefinger uden at klemme den for meget.
- Skub nålen helt ind. Din læge eller sygeplejerske har vist dig hvordan.
- Skub stemplet med tommelfingeren, indtil den fulde mængde væske, der svarer til den korrekte dosis, injiceres. Skub langsomt og jævnt, mens huden holdes hævet. PROTECS ™ nålesikringen aktiveres ikke, før injektionen er leveret. Dosis fuldt ud. Du hører muligvis et "klik", når PROTECS ™ nålesikringen er aktiveret.
- Når stemplet har nået slutningen af sit slag, skal du trække nålen ud og slippe huden.
- Fjern langsomt din tommelfinger fra stemplet, så nålen kan dækkes helt af sikkerhedsanordningen.
- I slutningen af injektionen kan der være en lille mængde blod på injektionsstedet. Dette er normalt. Du kan desinficere injektionsstedet ved at trykke på den antiseptiske pude i et par sekunder.
- Bortskaf brugte sprøjter i en sikker beholder - se afsnittet Opbevaring af Eprex
Overdosering Hvad skal jeg gøre, hvis du har taget for meget Eprex
Hvis du har brugt mere EPREX end du burde
Fortæl det straks til din læge eller sygeplejerske, hvis du mener, at du har brugt for meget EPREX. Bivirkninger fra overdosering er usandsynlige.
Hvis du har glemt at injicere EPREX
Udfør den næste injektion, så snart du husker det. Hvis den næste injektion falder inden for et døgn, skal du springe den glemte dosis over og fortsætte med det sædvanlige skema. Du må ikke fordoble injektionerne.
Hvis du er patient med hepatitis C og modtager interferon og ribavirin
Du bør kontakte din læge, fordi kombinationen af epoetin alfa med interferon og ribavirin i sjældne tilfælde har ført til tab af effekt og udviklingen af en alvorlig form for anæmi kaldet ren rød celle aplasi (PRCA). EPREX er ikke godkendt til behandling af anæmi forbundet med hepatitis C.
Spørg din læge, sygeplejerske eller apotek, hvis du har yderligere spørgsmål om brugen af dette produkt.
Bivirkninger Hvad er bivirkningerne af Eprex
Som al anden medicin kan EPREX forårsage bivirkninger, men ikke alle får bivirkninger. Fortæl det straks til din læge eller sygeplejerske, hvis nogen af nedenstående virkninger forekommer.
Meget almindelige bivirkninger
De forekommer hos mere end 1 ud af 10 patienter, der bruger EPREX
- Diarré
- Føler mig syg i maven
- Han trak sig tilbage
- Feber
- Overbelastning i luftvejene, såsom tilstoppet næse og ondt i halsen, er blevet rapporteret hos patienter med nyresygdom, der endnu ikke er under dialyse.
Almindelige bivirkninger
Disse påvirker op til 1 ud af 10 patienter, der bruger EPREX
- Forhøjelse af blodtryksværdier. Hovedpine (især ved pludselig, akut og migrænelignende) eller forvirring eller anfald. Dette kan være tegn på en pludselig stigning i blodtrykket, hvilket kræver øjeblikkelig behandling. Forøgelsen af blodtrykket kan kræve behandling. Med medicin (eller en dosisjustering af medicin, du allerede tager for forhøjet blodtryk).
- Blodpropper (herunder dyb venetrombose og emboli), som kan kræve akut behandling. Symptomer kan være brystsmerter, hvæsen, smertefuld hævelse af underekstremiteterne og rødme, normalt i benene.
- hoste.
- Hudirritation, der kan være forårsaget af en allergisk reaktion.
- Knogle- eller muskelsmerter.
- Influenzalignende syndrom, såsom hovedpine, smerter og smerter i leddene, følelse af svaghed, kulderystelser, træthed og svimmelhed. Disse reaktioner er mere almindelige i starten af behandlingen. Hvis du oplever disse symptomer, mens du injicerer i en vene, kan langsommere administration hjælpe med at undgå dem i fremtiden.
- Rødme, svie og smerter på injektionsstedet.
- Hævelse i ankler, fødder eller fingre.
Ikke almindelige bivirkninger
Disse påvirker op til 1 ud af 100 patienter, der bruger EPREX
- Høje niveauer af kalium i blodet, som kan forårsage en unormal hjerterytme (dette er en meget almindelig bivirkning hos dialysepatienter).
- Kramper
- Overbelastning af næsen eller luftvejene
Meget sjældne bivirkninger
Disse påvirker op til 1 ud af 10.000 patienter, der bruger EPREX
- Symptomer på Pure Red Cell Aplasia (PRCA). PRCA betyder manglende evne til at lave nok røde blodlegemer i knoglemarven. PRCA kan forårsage alvorlig og pludselig anæmi, hvis symptomer er:
- usædvanlig træthed,
- følelse af svimmelhed,
- åndenød.
PRCA er meget sjældent fundet, især hos patienter med nyresygdom efter måneder eller års behandling med EPREX og andre stoffer, der stimulerer produktionen af røde blodlegemer. En stigning i niveauet af små blodlegemer (kaldet blodplader), der normalt er involveret i blodproppdannelse, kaldet blodplader, kan forekomme, især i starten af behandlingen.Din læge vil kontrollere dette.
Hvis du er i hæmodialyse:
- Blodpropper (trombose) kan dannes i dialyse -shunten. Dette kan lettere ske, hvis du har lavt blodtryk (hypotension), eller hvis du har problemer med din fistel.
- Blodpropper kan også dannes i hæmodialysesystemet.Din læge kan beslutte at øge dosen heparin under dialyse.
Fortæl det straks til din læge eller sygeplejerske, hvis du bliver opmærksom på nogen af disse virkninger, eller hvis du bemærker andre virkninger, mens du tager EPREX.
Fortæl det til din læge, sygeplejerske eller apotek, hvis nogen af bivirkningerne bliver alvorlige, eller du bemærker nogen bivirkninger, der ikke er angivet i denne indlægsseddel.
For dem, der udøver sportsaktiviteter: brug af stoffet uden terapeutisk nødvendighed udgør doping og kan under alle omstændigheder afgøre positive antidopingtest.
Udløb og opbevaring
Opbevares utilgængeligt for børn.
Brug ikke dette lægemiddel efter den udløbsdato, der står på kartonen og etiketten efter bogstaverne EXP. Udløbsdatoen er den sidste dag i den angivne måned.
EPREX skal opbevares i køleskab (2 ° C - 8 ° C).
EPREX kan tages ud af køleskabet og opbevares ved stuetemperatur (ikke over 25 ° C) i op til 3 dage. Når den fyldte injektionssprøjte er fjernet fra køleskabet og har nået stuetemperatur (ikke over 25 ° C), skal den bruges inden for 3 dage eller kasseres.
Det må ikke fryses eller rystes
Opbevares i den originale emballage for at beskytte dem mod lys.
Brug ikke denne medicin, hvis forseglingen er brudt, eller hvis opløsningen er farvet eller suspenderede partikler observeres. Hvis disse forhold overholdes, skal medicinen kasseres
Lægemidler bør ikke bortskaffes via spildevand eller husholdningsaffald. Spørg din apotek om, hvordan du skal smide medicin, du ikke længere bruger. Dette vil hjælpe med at beskytte miljøet.
Pakningens indhold og andre oplysninger
Hvad indeholder Eprex
Den aktive ingrediens er: Epoetin alfa (mængden henvises til nedenstående tabel).
Øvrige indholdsstoffer er: Polysorbat 80, natriumchlorid, monobasisk natriumphosphatdihydrat, dibasisk natriumphosphatdihydrat, glycin og vand til injektionsvæsker. Denne medicin indeholder mindre end 1 mmol natrium (23 mg) pr. Dosis, så den er i det væsentlige natriumfri.
Hvordan EPREX ser ud og pakningens indhold
EPREX er en injektionsvæske, opløsning i fyldte sprøjter. De fyldte sprøjter er udstyret med en PROTECS ™ nålesikring (se tabellen nedenfor). EPREX er en klar og farveløs løsning.
Ikke alle pakningsstørrelser er nødvendigvis markedsført
Indlægsseddel: AIFA (Italian Medicines Agency). Indhold offentliggjort i januar 2016. De foreliggende oplysninger er muligvis ikke opdaterede.
For at få adgang til den mest opdaterede version er det tilrådeligt at få adgang til webstedet AIFA (Italian Medicines Agency). Ansvarsfraskrivelse og nyttige oplysninger.
01.0 LÆGEMIDLETS NAVN
EPREX 10000 IE / ML OPLØSNING TIL INJEKTION I FØRFyldt sprøjte
02.0 KVALITATIV OG KVANTITATIV SAMMENSÆTNING
Epoetin alfa 10.000 IE / ml (84,0 μg pr. Ml) fremstillet ved rekombinant DNA -teknologi i kinesiske hamsterovarier (CHO) celler.
En 0,3 ml fyldt injektionssprøjte indeholder 3000 IE (25,2 mcg) epoetin alfa
En 0,4 ml fyldt injektionssprøjte indeholder 4.000 IE (33,6 mcg) epoetin alfa
En 0,5 ml fyldt injektionssprøjte indeholder 5.000 IE (42,0 mcg) epoetin alfa
En 0,6 ml fyldt injektionssprøjte indeholder 6.000 IE (50,4 mikrogram) epoetin alfa
En 0,8 ml fyldt injektionssprøjte indeholder 8.000 IE (67,2 mcg) epoetin alfa
Én 1,0 ml fyldt injektionssprøjte indeholder 10.000 IE (84,0 mcg) epoetin alfa
Dette lægemiddel indeholder mindre end 1 mmol (23 mg) natrium pr. Dosis, dvs. det er i det væsentlige "natriumfrit".
Den fulde liste over hjælpestoffer findes i afsnit 6.1
03.0 LÆGEMIDDELFORM
Injektionsvæske, opløsning i fyldte sprøjter
Klar, farveløs løsning
04.0 KLINISKE OPLYSNINGER
04.1 Terapeutiske indikationer
EPREX er indiceret til behandling af symptomatisk anæmi forbundet med kronisk nyresvigt (CRI):
• hos voksne og pædiatriske patienter i alderen 1 - 18 år på hæmodialyse og hos voksne patienter i peritonealdialyse.
• hos voksne patienter med nyreinsufficiens, der endnu ikke er under dialyse til behandling af alvorlig anæmi af nyreoprindelse ledsaget af kliniske symptomer hos patienter.
EPREX er indiceret til voksne patienter, der modtager kemoterapi for solide tumorer, malignt lymfom eller myelomatose og med risiko for transfusion som angivet af patientens generelle status (kardiovaskulær situation, eksisterende anæmi ved kemoterapiens start) til behandling af anæmi og reduktion af behovstransfusionen.
EPREX er indiceret til voksne patienter, der er en del af et predonationsprogram for at øge mængden af autologt blod.Behandling er kun indiceret til patienter med moderat anæmi (hæmoglobinkoncentration i området Hb 10-13 g / dl [6, 2 - 8.1) mmol / l], ingen jernmangel), hvis blodlagringsprocedurer ikke er tilgængelige eller utilstrækkelige i tilfælde af større elektiv kirurgi, der kræver en stor mængde blod (4 eller flere enheder pr. kvinde eller 5 eller flere enheder for mænd).
EPREX er indiceret til voksne patienter, der ikke har jernmangel før større elektiv ortopædkirurgi, som menes at have stor risiko for transfusionskomplikationer, for at reducere eksponeringen for allogene blodtransfusioner. Brug bør begrænses. Til patienter med moderat anæmi (hæmoglobinkoncentration) i intervallet Hb10-13 g / dl), for hvem der ikke er et autologt blodprædonationsprogram, og for hvem der forventes moderat blodtab (fra 900 til 1800 ml).
04.2 Dosering og indgivelsesmåde
Dosering
Inden behandlingen påbegyndes med epoetin alfa, og når man beslutter at øge dosis, alle andre årsager til anæmi (jern-, folat- eller vitamin B12 -mangel, aluminiumforgiftning, infektioner eller betændelse, blodtab, hæmolyse og knoglemarvsfibrose af enhver oprindelse). For at sikre en optimal reaktion på epoetin alfa skal der sikres tilstrækkelige jernlagre og administreres jerntilskud om nødvendigt (se pkt. 4.4).
Behandling af symptomatisk anæmi hos voksne patienter med kronisk nyresvigt (CRI)
Symptomer og følgevirkninger af anæmi kan variere alt efter køn, alder og igangværende komorbiditeter; lægens vurdering af den enkelte patients kliniske tilstand er nødvendig.
Den ønskede hæmoglobinkoncentration er mellem 10 g / dl og 12 g / dl (6,2 til 7,5 mmol / l). EPREX bør administreres på en sådan måde, at der opnås en hæmoglobinkoncentration, der ikke overstiger 12 g / dl (7,5 mmol / l). En stigning i hæmoglobin større end 2 g / dl (1,25 mmol / l) over en fire-ugers periode bør undgås. Hvis dette sker, bør der foretages en passende dosisjustering.
På grund af variation i patienten kan hæmoglobinværdier over og under den ønskede hæmoglobinkoncentration lejlighedsvis observeres hos en patient. Denne variation skal håndteres gennem dosisjusteringer under hensyntagen til hæmoglobinkoncentrationsområdet mellem 10 g / dl (6,2 mmol / l) og 12 g / dl (7,5 mmol / l).
Et hæmoglobinniveau konsekvent over 12 g / dl (7,5 mmol) bør undgås. Hvis hæmoglobin stiger mere end 2 g / dL (1,25 mmol / L) pr. Måned, eller hvis hæmoglobinniveauet konsekvent overstiger 12 g / dL (7,5 mmol), reduceres EPREX -dosen med 25%. Hvis hæmoglobinet overstiger 13 g / dl ( 8,1 mmol / l), afbryd behandlingen, indtil den vender tilbage til 12 g / dl (7,5 mmol / l), og fortsæt derefter EPREX i doser, der er 25% lavere end de foregående.
Patienter bør overvåges nøje for at sikre, at den laveste tilladte effektive dosis EPREX bruges til at sikre tilstrækkelig kontrol af anæmi og relaterede symptomer ved at opretholde en hæmoglobinkoncentration under eller lig med 12 g / dl (7, 5 mmol / l).
Der bør udvises forsigtighed ved at øge ESA -dosis til patienter med kronisk nyresvigt.For patienter med dårligt hæmoglobinrespons på ESA'er bør der søges en alternativ årsag til den dårlige respons (se pkt. 4.4 og 5.1).
EPREX -behandling er opdelt i to faser - korrektionsfase og vedligeholdelsesfase.
Voksne patienter i hæmodialyse
Hos hæmodialysepatienter, hvor venøs adgang er let tilgængelig, foretrækkes anvendelse af den intravenøse indgivelsesvej.
Korrektionsfase :
Startdosis er 50 IE / kg vægt 3 gange om ugen.
Hvis det er nødvendigt, øges eller sænkes dosis med 25 IE / kg (3 gange om ugen), indtil den ønskede hæmoglobinkoncentration er i intervallet 10g / dl til 12g / dl (6,2 til 7,5 mmol / l) (dette skal ske gradvist kl. mindst fire uger).
- Vedligeholdelsesfase :
Den anbefalede samlede ugentlige dosis er mellem 75 IE / kg og 300 IE / kg.
Der bør foretages passende dosisjustering for at opretholde hæmoglobinværdier inden for den ønskede hæmoglobinkoncentration mellem 10 g / dl og 12 g / dl (6,2 til 7,5 mmol / l).
Patienter med et meget lavt indledende hæmoglobinniveau (8 g / dl eller> 5 mmol / l).
Voksne patienter med nyreinsufficiens, der endnu ikke er i dialyse
Hos patienter, hvor venøs adgang ikke er let tilgængelig, kan EPREX administreres subkutant.
Korrektionsfase
Startdosis er 50 IE / kg vægt, 3 gange om ugen efterfulgt om nødvendigt af en dosisforøgelse på 25 IE / kg (3 gange om ugen), indtil den ønskede hæmoglobinkoncentration er nået (dette skal gøres gradvist. Kl. mindst fire uger).
Vedligeholdelsesfase
Under vedligeholdelsesfasen kan EPREX administreres enten 3 gange om ugen og, i tilfælde af subkutan administration, en gang om ugen eller en gang hver anden uge.
Dosis og doseringsintervaller skal justeres korrekt for at opretholde hæmoglobinværdier på det ønskede niveau: Hb mellem 10 og 12 g / dl (6,2-7,5 mmol / l). Forlængelse af doseringsintervallet kan kræve en dosisforøgelse.
Den maksimale dosis bør ikke overstige 150 IE / kg 3 gange om ugen, 240 IE / kg (op til maksimalt 20.000 IE) en gang om ugen eller 480 IE / kg (op til et maksimum på 40.000 IE) en gang hver anden uge.
Voksne patienter i peritonealdialyse
Hos patienter, hvor venøs adgang ikke er let tilgængelig, kan EPREX administreres subkutant.
Korrektionsfase
Startdosis er 50 IE / kg, to gange om ugen.
Vedligeholdelsesfase
Den anbefalede vedligeholdelsesdosis er mellem 25 IE / kg og 50 IE / kg, to gange om ugen, opdelt i 2 lige administrationer.
Der bør foretages passende dosisjustering for at opretholde hæmoglobinværdier på det ønskede niveau: hæmoglobin mellem 10 g / dl og 12 g / dl (6,2-7,5 mmol / l).
Behandling af voksne patienter med kemoterapi-induceret anæmi
Symptomerne og følgevirkningerne af anæmi kan variere alt efter alder, køn og sygdomens generelle situation; lægens vurdering af den enkelte patients kliniske tilstand er nødvendig.
EPREX bør administreres til anæmiske patienter f.eks. hæmoglobinkoncentration ≤10 g / dl (6,2 mmol / l).
Startdosis er 150 IE / kg, administreret subkutant, 3 gange om ugen.
Alternativt kan EPREX administreres subkutant ved startdosis på 450 IE / kg en gang ugentligt.
Der bør foretages passende dosisjustering for at opretholde hæmoglobinværdier på det ønskede niveau: hæmoglobin mellem 10 g / dl og 12 g / dl (6,2-7,5 mmol / l).
På grund af variation i patienten kan hæmoglobinværdier over og under den ønskede hæmoglobinkoncentration lejlighedsvis observeres hos en patient. Denne variabilitet skal håndteres gennem dosisjusteringer under respekt for det ønskede hæmoglobinkoncentrationsinterval mellem 10 g / dl (6,2 mmol / l) og 12 g / dl (7,5 mmol / l).
En hæmoglobinkoncentration konsekvent over 12 g / dl (7,5 mmol) bør undgås. Instruktioner til korrekt dosisjustering, hvis hæmoglobin når koncentrationer over 12 g / dl (7,5 mmol), er angivet nedenfor.
Hvis hæmoglobinkoncentrationen efter 4 ugers behandling er steget med mindst 1 g / dL (0,62 mmol / L), eller antallet af reticulocytter er steget med ≥ 40.000 celler / μl fra baseline, bør dosis forblive på 150 IE / kg 3 gange om ugen eller 450 IE / kg en gang om ugen.
Hvis stigningen i hæmoglobinkoncentrationen er
Hvis stigningen i hæmoglobinkoncentrationen var
Dosisjustering for at opretholde hæmoglobinkoncentrationer mellem 10 g / dl - 12 g / dl
Hvis hæmoglobinkoncentrationen stiger mere end 2 g / dl (1,25 mmol / l) pr. Måned, eller hvis hæmoglobinet overstiger 12 g / dl (7,5 mmol / l), reduceres dosis af EPREX på ca. 25-50%.
Hvis hæmoglobinkoncentrationen overstiger 13 g / dl (8,1 mmol / l), skal behandlingen afbrydes, indtil koncentrationen falder til under 12 g / dl (7,5 mmol / l), og derefter genoptages behandlingen med EPREX i en dosis, der er 25% lavere end den tidligere dosis.
Den anbefalede dosis er beskrevet i følgende diagram:
Patienter bør overvåges nøje for at sikre, at den laveste godkendte dosis af erytropoiesestimulerende midler (ESA) bruges til at give tilstrækkelig kontrol med symptomerne på anæmi.
EPREX -behandlingen bør fortsættes i en måned efter afslutningen af kemoterapien.
Behandling af voksne patienter, der er kandidater til kirurgi, der er en del af et autologt blodprononationsprogram
Mildt anæmiske patienter (hæmatokrit mellem 33-39%), der kræver foruddeponering af 4 eller flere enheder blod, skal behandles med 600 IE / kg EPREX intravenøst, to gange om ugen, i løbet af de 3 uger før operationen.
EPREX bør administreres efter afslutningen af bloddonationsproceduren.
Behandling af voksne patienter kandidater til større elektiv ortopædkirurgi
Den anbefalede dosis er 600 IE / kg EPREX administreret subkutant, en gang om ugen i løbet af de tre uger før operationen (-21 dage, -14 dage og -7 dage) og på operationsdagen.
Hvis der er et medicinsk behov for at reducere ventetiden før operationen til mindre end tre uger, skal dosis på 300 IE / kg EPREX administreres subkutant dagligt i 10 på hinanden følgende dage. Før operationen, på operationsdagen og i de 4 dage umiddelbart efter det.
Hvis hæmoglobin når 15 g / dl eller mere i den præoperative periode, bør administrationen af EPREX afbrydes, og der bør ikke administreres yderligere doser.
Pædiatrisk population
Behandling af symptomatisk anæmi hos patienter med kronisk nyresvigt ved hæmodialyse
Symptomer og følgevirkninger af anæmi kan variere alt efter alder, køn og igangværende komorbiditeter; lægens vurdering af den enkelte patients kliniske tilstand er nødvendig.
Hos pædiatriske patienter er den ønskede hæmoglobinkoncentration mellem 9,5 g / dl og 11 g / dl (5,9 til 6,8 mmol / l). EPREX bør administreres på en sådan måde, at der opnås en hæmoglobinkoncentration på højst 11 g / dl (6,8 mmol / l). En stigning i hæmoglobin større end 2 g / dl (1,25 mmol / l) over en fire-ugers periode bør undgås. Hvis dette sker, bør der foretages en passende dosisjustering.
Patienter bør overvåges nøje for at sikre, at den laveste autoriserede dosis EPREX bruges til at sikre tilstrækkelig kontrol af anæmi og relaterede symptomer.
EPREX -behandling er opdelt i to faser: korrektionsfase og vedligeholdelsesfase.
Hos pædiatriske patienter i hæmodialyse, hvor intravenøs adgang allerede er tilgængelig, foretrækkes intravenøs administration.
Korrektionsfase:
Startdosis er 50 IE / kg vægt intravenøst, 3 gange om ugen.
Hvis det er nødvendigt, øges eller reduceres dosis med 25 IE / kg (3 gange om ugen) indtil den ønskede hæmoglobinkoncentration i området 9,5 g / dl til 11 g / dl (5,9 til 6, 8 mmol / l) (dette bør ske gradvist med mindst fire ugers mellemrum).
Vedligeholdelsesfase :
Der bør foretages passende dosisjustering for at opretholde hæmoglobinværdier inden for den ønskede hæmoglobinkoncentration mellem 9,5 g / dl og 11 g / dl (5,9 til 6,8 mmol / l).
Generelt kræver børn, der vejer mindre end 30 kg, højere vedligeholdelsesdoser end børn, der vejer mere end 30 kg og voksne.
Pædiatriske patienter med et meget lavt indledende hæmoglobinniveau (6,8 g / dL eller> 4,25 mmol / L).
Behandling af pædiatriske patienter med kemoterapiinduceret anæmi.
Sikkerheden og effekten af EPREX hos pædiatriske patienter, der får kemoterapi, er ikke fastslået.
Behandling af pædiatriske kirurgiske patienter, der deltager i et autologt predonationsprogram
Sikkerheden og effekten af EPREX hos pædiatriske patienter er ikke fastslået.
Ingen data er tilgængelige.
Behandling af pædiatriske patienter, der afventer større elektiv ortopædkirurgi
Sikkerheden og effekten af EPREX hos pædiatriske patienter er ikke fastslået.
Ingen data er tilgængelige.
Indgivelsesmåde
Vær forsigtig, før du håndterer eller administrerer medicinen.
Før brug, lad EPREX -sprøjten hvile, indtil den når stuetemperatur. Dette tager normalt mellem 15 og 30 minutter.
Behandling af symptomatisk anæmi hos voksne patienter med kronisk nyresvigt
Hos patienter med kronisk nyresvigt, hvor der normalt er intravenøs adgang (patienter med hæmodialyse), foretrækkes intravenøs administration af EPREX.
Når intravenøs adgang ikke er let tilgængelig (patienter, der endnu ikke er i dialyse og patienter i peritonealdialyse), kan EPREX administreres subkutant.
Behandling af voksne patienter med kemoterapi-induceret anæmi.
EPREX skal administreres subkutant.
Behandling af voksne kirurgiske patienter, der deltager i et autologt predonationsprogram
EPREX skal administreres intravenøst.
Behandling af voksne patienter planlagt til større elektiv ortopædkirurgi
EPREX skal administreres subkutant.
Behandling af symptomatisk anæmi hos pædiatriske patienter med kronisk nyresvigt ved hæmodialyse
Hos pædiatriske patienter med kronisk nyresvigt, hvor intravenøs adgang allerede er tilgængelig (hæmodialysepatienter), foretrækkes intravenøs administration af EPREX.
Intravenøs administration
Administration bør tage mindst 1-5 minutter afhængigt af den samlede dosis.
Hos hæmodialysepatienter kan bolusinjektionen gives under dialyse gennem tilstrækkelig venøs adgang i dialyselinjen Alternativt kan injektionen gives ved dialysens afslutning, gennem adgangen til fistlen, efterfulgt af administration af 10 ml af fysiologisk løsning for at skylle adgangsvejene og sikre en tilfredsstillende introduktion af produktet i blodbanen.
Hos patienter, der har oplevet influenzalignende reaktioner, foretrækkes langsommere administration (se pkt. 4.8).
Administrer ikke EPREX som intravenøs infusion eller i opløsning med andre lægemidler.
Subkutan administration
Det maksimale volumen på 1 ml på hvert injektionssted bør generelt ikke overskrides. Ved større mængder skal der vælges mere end et injektionssted.
Injektioner skal foretages i lemmerne eller den forreste mavevæg.
Hvis lægen mener, at patienten eller omsorgspersonen er i stand til at administrere EPREX sikkert og hensigtsmæssigt subkutant, bør der gives instruktioner om korrekt dosis og administration.
Som med andre injicerbare produkter skal du kontrollere, at der ikke er partikler i opløsningen eller farvevariationer.
04.3 Kontraindikationer
Overfølsomhed over for det aktive stof eller over for et eller flere af hjælpestofferne.
Patienter, der udvikler Pure Red Cell Aplasia (PRCA) efter behandling med erythropoietin, bør ikke behandles med EPREX eller andre erythropoietiner (se pkt. 4.4 PRCA).
Ukontrolleret hypertension.
Alle kontraindikationer forbundet med det autologe blod-deponeringsprogram skal huskes på patienter behandlet med EPREX.
Anvendelse af EPREX er kontraindiceret ved tilstedeværelse af alvorlige vaskulære lidelser på koronar, perifer arteriel, carotis eller cerebralt niveau hos patienter, der er kandidater til større elektiv ortopædkirurgi og ikke er en del af et autologt predonationsprogram. Brugen er også kontraindiceret. hos patienter med nylige episoder med myokardieinfarkt eller andre cerebro-vaskulære komplikationer.
Patienter, der er kandidater til kirurgi, som af en eller anden grund ikke kan modtage tilstrækkelig antitrombotisk profylakse.
04.4 Særlige advarsler og passende forholdsregler ved brug
Generel
Hos alle patienter, der får epoetin alfa, skal blodtrykket overvåges nøje og kontrolleres efter behov. Epoetin alfa bør bruges med forsigtighed i nærvær af ubehandlet, utilstrækkeligt behandlet eller svært at kontrollere hypertension. Antihypertensiv behandling skal muligvis påbegyndes eller intensiveres. Hvis blodtrykket ikke kan kontrolleres, skal behandlingen med epoetin alfa afbrydes.
Hypertensive kriser med encefalopati og anfald, der kræver øjeblikkelig lægehjælp og intensiv lægehjælp, er opstået under behandling med epoetin alfa, selv hos patienter med tidligere normalt eller lavt blodtryk. Særlig opmærksomhed bør rettes mod migrænelignende twinges som et muligt advarselsskilt (se afsnit 4.8).
Epoetin alfa bør anvendes med forsigtighed til patienter med epilepsi, anfald i anfald eller medicinske tilstande forbundet med disposition for anfaldsaktivitet såsom infektioner i centralnervesystemet og hjernemetastaser.
Epoetin alfa bør anvendes med forsigtighed til patienter med kronisk leversvigt.Sikkerheden ved epoetin alfa er ikke fastslået hos patienter med nedsat leverfunktion.
En øget forekomst af vaskulære trombotiske hændelser (VTE'er) er blevet observeret hos patienter, der har modtaget ESA'er (se pkt. 4.8) Disse omfatter venøs og arteriel trombose og emboli (herunder nogle med dødelig udgang) såsom dyb venetrombose, lungeemboli, nethinde trombose og myokardieinfarkt Derudover er der rapporteret cerebrovaskulære ulykker (herunder hjerneinfarkt, hjerneblødning og forbigående iskæmiske anfald).
Risikoen for disse VTE'er bør omhyggeligt afvejes mod fordelene ved behandling med epoetin alfa, især hos patienter med allerede eksisterende risikofaktorer for VTE, herunder fedme og tidligere VTE (f.eks. Dyb venetrombose, lungeemboli og cerebrovaskulære ulykker)
Hæmoglobinniveauer bør overvåges nøje hos alle patienter på grund af en potentiel øget risiko for tromboemboliske hændelser og fatale følger, når patienter behandles med hæmoglobinniveauer over den angivne koncentration.
En moderat dosisafhængig stigning i antallet af blodplader kan forekomme under behandling med epoetin alfa, dog inden for det normale område. Dette fænomen går tilbage i løbet af terapien. Derudover er der rapporteret om trombocytæmi over det normale område.Det anbefales, at blodplader overvåges regelmæssigt i løbet af de første 8 uger af behandlingen.
Alle mulige årsager til anæmi (jernmangel, folat- eller vitamin B12 -mangel, aluminiumforgiftning, infektion eller betændelse, blodtab, hæmolyse og knoglemarvsfibrose af enhver oprindelse) bør vurderes og behandles, inden behandlingen påbegyndes. Med epoetin alfa og når der træffes en beslutning er lavet for at øge dosis. I de fleste tilfælde falder serumferritinværdierne samtidig med, at hæmatokritværdierne stiger.For at sikre en optimal reaktion på epoetin alfa bør der sikres tilstrækkelige jernlagre, og om nødvendigt bør jerntilskud gives ( se afsnit 4.2):
• Til patienter med kronisk nyresvigt anbefales jerntilskud, hvis ferritinniveauerne er under 100 ng / ml (elementært jern til voksne 200 til 300 mg / dag oralt og til børn fra 100 til 200 mg / dag oralt).
• For kræftpatienter anbefales jerntilskud, hvis værdierne for transferrinmætning er mindre end 20% (elementært jern 200 til 300 mg / dag oralt).
• For patienter i et autologt predonationsprogram bør jerntilskud (elementært jern 200 mg / dag oralt) administreres et par uger før starten på autolog predonation for at opnå høje jernlagre. Inden behandlingens start med epoetin alfa og under forløbet af behandling med epoetin alfa.
• For patienter, der er planlagt til større elektiv ortopædkirurgi, bør jerntilskud (elementært jern 200 mg / dag oralt) administreres under epoetin alfa -behandling. "Jerntilskud, inden epoetin alfa -behandlingen påbegyndes for at opnå en tilstrækkelig jernreserve.
Begyndelse eller forværring af porfyri er meget sjældent observeret hos patienter behandlet med epoetin alfa.
Epoetin alfa bør bruges med forsigtighed til patienter med porfyri.
For at sikre sporbarheden af Erythropoiesis Stimulating Agents (ESA) skal det administrerede ESA's kommercielle navn altid registreres eller angives i patientens journal.
Ændring af terapi fra en ESA til en anden bør kun foretages under passende overvågning.
Pure Red Cell Aplasia (PRCA)
Antistof-medieret Pure Red Cell Aplasia (PRCA) er blevet rapporteret efter måneder til år, hovedsageligt hos patienter med kronisk nyresvigt behandlet med epoetin administreret subkutant.
Tilfælde er også blevet rapporteret hos patienter med hepatitis C behandlet med interferon og ribavirin, når de gives i kombination med ESA. Epoetin alfa er ikke godkendt til behandling af anæmi forbundet med hepatitis C.
Hos patienter, der udviser et pludseligt tab af effekt, defineret som et fald i hæmoglobinværdier (1 til 2 g / dl pr. Måned) med et øget behov for transfusioner, bør der foretages et antal retikulocytter og evalueres kendte årsager. Som forhindrer reaktion (såsom jern-, folat- og vitamin B12 -mangel, aluminiumforgiftning, infektion eller betændelse, blodtab, hæmolyse og knoglemarvsfibrose af enhver oprindelse).
Et uforholdsmæssigt fald i hæmoglobinværdier og udvikling af alvorlig anæmi forbundet med lavt antal reticulocytter bør føre til afbrydelse af behandlingen med epoetin alfa og udføre en test for tilstedeværelsen af anti-erythropoietin-antistoffer.
En knoglemarvsundersøgelse til diagnose af PRCA bør også overvejes.
Ingen behandling med andre ESA'er bør påbegyndes på grund af risikoen for krydsreaktion.
Behandling af symptomatisk anæmi hos voksne og pædiatriske patienter med kronisk nyresvigt (CRI):
Patienter med kronisk nyresvigt, der modtager epoetin alfa, bør have deres hæmoglobinniveauer målt regelmæssigt, indtil et stabilt niveau er nået og derefter måles periodisk derefter.
Hos patienter med kronisk nyresvigt for at reducere risikoen for blodtryksstigninger, bør hæmoglobin stige med cirka 1 g / dl / måned (0,62 mmol / l) og må ikke overstige 2 g / dl / måned (1,25 mmol / L).
Hos patienter med kronisk nyreinsufficiens bør vedligeholdelseshæmoglobinkoncentrationen ikke overstige den maksimale værdi af hæmoglobinkoncentrationsområdet som rapporteret i afsnit 4.2 Der er observeret en øget risiko for dødelighed og alvorlige kardiovaskulære hændelser i kliniske forsøg, når erytropoiesestimulerende midler (ESA'er) var administreres for at opnå et hæmoglobinkoncentrationsniveau over 12 g / dl (7,5 mmol / ml).
Kontrollerede kliniske undersøgelser har ikke vist nogen væsentlig fordel ved administration af epoetiner, når hæmoglobinkoncentrationen overstiger det niveau, der er nødvendigt for at kontrollere symptomerne på anæmi og for at undgå blodtransfusioner.
Der bør udvises forsigtighed ved at øge dosis af EPREX til patienter med kronisk nyresvigt, da høje kumulative doser af epoetin kan være forbundet med en øget risiko for dødelighed, alvorlige kardiovaskulære og cerebrovaskulære hændelser.For patienter med dårlig hæmoglobinrespons. På epoetiner, et alternativ årsag til denne dårlige reaktion bør søges (se afsnit 4.2 og 5.1).
Patienter med kronisk nyresvigt behandlet med subkutan epoetin alfa bør monitoreres regelmæssigt for tab af effekt, hvilket betyder fravær eller nedsat respons på epoetin alfa hos patienter, der tidligere reagerede på behandlingen. Dette fænomen er kendetegnet ved et vedvarende fald i hæmoglobinværdier sammenlignet med en stigning i dosis af epoetin alfa (se pkt. 4.8).
Nogle patienter, der behandles med epoetin alfa med længere doseringsintervaller (større end en gang om ugen), bevarer muligvis ikke tilstrækkelige hæmoglobinniveauer (se pkt. 5.1) og kan kræve en dosisforøgelse. Hæmoglobinniveauer bør overvåges regelmæssigt.
Trombose af vaskulær adgang er forekommet hos patienter i hæmodialyse, især hos patienter med tendens til hypotension og med komplikationer af arteriovenøse fistler (f.eks. Stenose, aneurismer osv.). Hos disse patienter anbefales forebyggende kontrol af vaskulær adgang og profylakse med trombose med administration af fx acetylsalicylsyre.
Hyperkaliæmi er blevet observeret i isolerede tilfælde, selvom årsagssammenhæng ikke er fastslået. Serumelektrolytter bør monitoreres hos patienter med kronisk nyresvigt. Hvis der observeres forhøjede (eller stigende) serumkaliumniveauer, ud over passende behandling af hyperkaliæmi, bør det overvejes at afbryde administrationen af epoetin alfa, indtil serumkaliumniveauer er blevet korrigeret.
Ofte under hæmodialyse er en stigning i heparindosis påkrævet på grund af stigningen i hæmatokritværdien. Hvis justeringen af heparindoser ikke er optimal, kan okklusion af dialysatoren forekomme. Baseret på de hidtil tilgængelige data fremskynder korrektionen af anæmi med epoetin alfa hos voksne patienter med kronisk nyresvigt, der endnu ikke er under dialyse, ikke udviklingen af nyresvigt.
Behandling af patienter med kemoterapi-induceret anæmi
Kræftpatienter, der modtager epoetin alfa, skal have deres hæmoglobinniveauer målt regelmæssigt, indtil et stabilt niveau er nået og derefter måles periodisk derefter.
Erythropoietiner er vækstfaktorer, der i det væsentlige stimulerer produktionen af røde blodlegemer. Erythropoietin -receptorer kan udtrykkes på overfladen af en række kræftceller. Som med alle vækstfaktorer er der teoretisk bekymring for, at erythropoietiner kan stimulere tumorvækst.
I nogle kontrollerede kliniske forsøg har erythropoietiner ikke vist sig at forbedre den samlede overlevelse eller reducere risikoen for tumorprogression hos patienter med tumorassocieret anæmi.
I kontrollerede kliniske forsøg har brugen af epoetin alfa og andre erytropoiesestimulerende midler (ESA'er) vist:
• reduceret lokalregionel kontrol hos patienter med fremskreden hoved- og halskræft, der modtager strålebehandling, når de administreres for at opnå hæmoglobinniveauer over 14 g / dl (8,7 mmol / l);
• reduceret samlet overlevelse og øget dødelighed, der kan tilskrives sygdomsprogression efter 4 måneder hos patienter med metastatisk brystkræft, der får kemoterapi, når de administreres for at opnå hæmoglobinniveauer mellem 12-14 g / dl (7,5-8,7 mmol / L);
• øget risiko for dødelighed ved administration for at opnå hæmoglobinniveauer på 12 g / dl (7,5 mmol / l); hos patienter med aktiv neoplasi, der ikke gennemgår kemo- og / eller strålebehandling. Behandling med erytropoiesestimulerende midler (ESA'er) er ikke indiceret i denne patientpopulation.
I lyset af ovenstående bør blodtransfusion foretrækkes i nogle kliniske situationer til behandling af anæmi hos kræftpatienter Beslutningen om at administrere rekombinant erythropoietin bør være baseret på en fordel-risiko-vurdering med individuel involvering. Af patienten, som skal overveje hans eller hendes særlige kliniske kontekst Faktorer, der skal overvejes under denne evaluering, skal omfatte kræftformen og dens fremskridt; graden af anæmi forventet levetid, miljøet, hvor patienten behandles; patientens præferencer (se afsnit 5.1).
Hos kræftpatienter, der modtager kemoterapi, bør intervallet på 2-3 uger mellem administration og fremkomsten af ESA-inducerede røde blodlegemer nøje overvejes, når det vurderes, om epoetin alfa-behandling er hensigtsmæssig (patienter med risiko for transfusion).
Voksne patienter, der er kandidater til kirurgiske indgreb, der er en del af autologe blodprædonationsprogrammer
Alle advarsler og særlige forholdsregler i forbindelse med det autologe blodprononationsprogram, især rutinemæssig volumenudskiftning, skal overholdes.
Patienter kandidater til større elektiv ortopædkirurgi
God blodhåndteringspraksis bør altid følges præoperativt.
Patienter, der er kandidater til valgfri større ortopædkirurgi, bør modtage "tilstrækkelig antitrombotisk profylakse, da der kan forekomme trombotiske og vaskulære hændelser hos patienter, der opereres, især hos dem med underliggende kardiovaskulære lidelser. Desuden skal der tages særlige forholdsregler hos patienter. Med disposition for udviklingen af dyb venetrombose (DVT). Desuden kan operatører ikke udelukkes hos patienter med et "baseline hæmoglobin> 13 g / dl", at behandling med epoetin alfa kan være forbundet med en øget risiko for posttrombotiske / vaskulære hændelser. . Epoetin alfa bør derfor ikke anvendes til patienter med baseline hæmoglobin> 13 g / dl.
04.5 Interaktioner med andre lægemidler og andre former for interaktion
Der er ingen tegn på, at behandling med epoetin alfa ændrer metabolismen af andre lægemidler. Lægemidler, der reducerer erythropoiesis, kan nedsætte responsen på epoetin alfa.
Da cyclosporin binder sig til røde blodlegemer, kan der være en "interaktion med dette lægemiddel.
I tilfælde af samtidig administration bør ciclosporins blodniveauer overvåges og dosis justeres i henhold til stigningen i hæmatokrit.
Der er ingen in vitro-interaktioner mellem G-CSF, GM-CSF og epoetin alfa vedrørende hæmatologisk differentiering eller proliferation in vitro af tumorbiopsiprøver.
Hos voksne kvindelige patienter med metastatisk brystkræft havde subkutan samtidig administration af 40.000 IE / ml epoetin alfa og trastuzumab 6 mg / kg ingen effekt på trastuzumabs farmakokinetik.
04.6 Graviditet og amning
Graviditet
Der er ingen tilstrækkelige og velkontrollerede undersøgelser af gravide. Dyrestudier har vist reproduktionstoksicitet (se pkt. 5.3). Følgelig bør epoetin alfa kun bruges under graviditet, hvis den forventede fordel opvejer den potentielle risiko for fosteret. Brug af epoetin alfa anbefales ikke til gravide kvinder, der er kandidater til kirurgi, der deltager i et autologt blodprononationsprogram.
Fodringstid
Det vides ikke, om eksogent epoetin alfa udskilles i human modermælk. Epoetin alfa bør bruges med forsigtighed til ammende kvinder.En beslutning om at fortsætte / afbryde amning eller at fortsætte / afbryde behandling med epoetin alfa skal tages i betragtning af fordelen ved amning for spædbarnet og fordelen ved epoetin alfa -behandling for kvinden.
Anvendelse af epoetin alfa anbefales ikke til ammende kvinder, der er kandidater til kirurgi, der deltager i et autologt blodprononationsprogram.
Fertilitet
Der er ingen undersøgelser, der vurderer den potentielle effekt af epoetin alfa på fertiliteten hos mænd eller kvinder.
04.7 Virkninger på evnen til at føre motorkøretøj og betjene maskiner
Der er ikke udført undersøgelser af evnen til at føre motorkøretøj eller betjene maskiner.
04.8 Bivirkninger
Resumé af sikkerhedsprofilen
Den hyppigste bivirkning under behandling med epoetin alfa er en dosisafhængig stigning i blodtryk eller forværring af eksisterende hypertension.
Det anbefales at overvåge blodtryksudviklingen, især i starten af behandlingen (se pkt.4.4).
De hyppigste bivirkninger, der forekom i kliniske forsøg med epoetin alfa, er diarré, kvalme, opkastning, feber og hovedpine. Influenzalignende symptomer kan hovedsageligt opstå i begyndelsen af behandlingen.
Overbelastning af luftvejene, herunder hændelser i overbelastning i øvre luftveje, nasal overbelastning og rhino-pharyngitis, er blevet rapporteret i kliniske forsøg med dosisudvidelser hos voksne patienter med nyreinsufficiens, der endnu ikke er under dialyse.
En øget forekomst af vaskulære trombotiske hændelser (TVE) er blevet observeret hos patienter, der fik ESA (se pkt. 4.4).
Tabel over bivirkninger
Af i alt 3.262 patienter i 23 randomiserede, dobbeltblinde, placebo- eller standardbehandlingskontrollerede kliniske forsøg blev den samlede sikkerhedsprofil af EPREX evalueret hos 1.992 anæmiske patienter. I 4 undersøgelser med kronisk nyresvigt blev 228 ICR-patienter behandlet med epoetin alfa inkluderet (2 studier i præ-dialyse [N = 131 ICR-eksponerede patienter] og 2 på dialyse [N = 97 ICR-eksponerede patienter]); 1.404 kræftpatienter udsat i 16 undersøgelser af anæmi forårsaget af kemoterapi; 147 patienter udsat i 2 undersøgelser for præ-donation af autologt blod og 213 patienter, der blev eksponeret i 1 undersøgelse i den perioperative periode. Bivirkninger rapporteret for ≥1% af patienterne behandlet med epoetin alfa i disse kliniske forsøg er vist i nedenstående tabel.
Følgende definitioner gælder for de forskellige frekvenser: meget almindelig (≥ 1/10), almindelig (≥ 1/100 til
Beskrivelse af udvalgte bivirkninger
Overfølsomhedsreaktioner, herunder tilfælde af udslæt (herunder urticaria), anafylaktiske reaktioner og angioødem er blevet rapporteret.
Hypertensive kriser med encefalopati og anfald, som krævede øjeblikkelig lægehjælp og intensiv lægehjælp, er opstået under behandling med epoetin alfa selv hos patienter med tidligere normalt eller lavt blodtryk. Der bør lægges særlig vægt på migrænelignende twinges som et muligt advarselstegn (se pkt. 4.4).
Antistof-medieret ren røde blodlegemer er blevet rapporteret meget sjældent i
Pædiatrisk population med kronisk nyresvigt ved hæmodialyse
Eksponeringen af pædiatriske patienter med kronisk nyresvigt i hæmodialyse i kliniske forsøg og efter markedsføring er begrænset. Der blev ikke rapporteret om specifikke bivirkninger fra den pædiatriske population, der ikke er nævnt i ovenstående tabel, eller bivirkninger, der er typiske for den underliggende sygdom, i denne population.
Indberetning af formodede bivirkninger
Rapportering af formodede bivirkninger, der opstår efter godkendelse af lægemidlet, er vigtig, da det muliggør kontinuerlig overvågning af lægemidlets fordel / risiko -balance. Sundhedspersonale anmodes om at rapportere alle formodede bivirkninger via det nationale rapporteringssystem. "Adresse http: //www.agenziafarmaco.gov.it/it/responsabili.
04.9 Overdosering
Den terapeutiske margen for epoetin alfa er meget bred. Overdosering med epoetin alfa kan forårsage effekter, der er forlængelser af hormonets farmakologiske virkninger. Flebotomi kan udføres, hvis der findes for høje hæmoglobinniveauer.
Yderligere understøttende pleje bør ydes som krævet af patientens tilstand.
05.0 FARMAKOLOGISKE EGENSKABER
05.1 Farmakodynamiske egenskaber
Farmakoterapeutisk gruppe: anti-anæmisk, ATC-kode: B03XA01.
Handlingsmekanisme
Erythropoietin (EPO) er et glycoproteinhormon, der primært produceres af nyrerne som reaktion på hypoxi og er nøgleregulatoren for produktionen af røde blodlegemer (RBC). "EPO er involveret i alle faser af erythroidudvikling og har sin hovedvirkning på niveauet af erythroid -forstadier. Efter at EPO har bundet til sin receptor på celleoverfladen, aktiverer den signaltransduktionsveje, der forstyrrer" apoptose og stimulerer erythroidcelleproliferation. Rekombinant human erythropoietin (epoetin alfa), udtrykt i ovarieceller fra kinesisk hamster, har en sekvens på 165 aminosyrer, der er identisk med menneskelig urin EPO; de 2 er ikke til at skelne på grundlag af funktionelle assays Den tilsyneladende molekylvægt af erythropoietin er mellem 32.000 og 40.000 dalton.
Erythropoietin er en vækstfaktor, der primært stimulerer produktionen af erythrocytter. Erythropoietin -receptorer kan udtrykkes på overfladen af en række kræftceller.
Farmakodynamiske virkninger
Sunde frivillige
Efter enkeltdoser af epoetin alfa (20.000 til 160.000 IE subkutant) blev der observeret en dosisafhængig respons for undersøgte farmakodynamiske markører, herunder: reticulocytter, røde blodlegemer og hæmoglobin. En defineret koncentration-tidsmæssig profil blev observeret med peak og tilbagevenden til baseline for ændringer i procentdelen af reticulocytter. En mindre defineret profil blev observeret for erythrocytter og hæmoglobin. Generelt steg alle farmakodynamiske markører lineært med doser, der nåede maksimal respons ved maksimale dosisniveauer.
Yderligere farmakodynamiske undersøgelser undersøgte 40.000 IE en gang om ugen sammenlignet med 150 IE / kg 3 gange om ugen. På trods af forskellene i koncentrationstidsprofiler var den farmakodynamiske respons (målt ved procentvise ændringer i reticulocytter, hæmoglobin og totale røde blodlegemer) ens for disse regimer. Yderligere undersøgelser sammenlignede 40.000 IE -regimet af epoetin alfa en gang ugentligt med doser hver uge fra 80.000 til 120.000 IE subkutant. Baseret på resultaterne af disse farmakodynamiske undersøgelser hos raske forsøgspersoner ser det generelt ud til, at doseringsregimet på 40.000 IE én gang ugentligt er mere effektivt i produktionen af røde blodlegemer end de to ugers behandlinger på trods af en lighed i produktionen af retikulocytter. En gang om ugen og to ugentlige regimer.
Kronisk nyresvigt
Epoetin alfa har vist sig at stimulere erythropoiesis hos anæmiske patienter med kronisk nyresvigt, herunder dialyse og præ-dialyse patienter.Det første tegn på et svar på epoetin alfa er en stigning i antallet af reticulocytter inden for 10 dage efterfulgt af stigninger i antallet af røde blodlegemer, hæmoglobin og hæmatokrit, normalt inden for 2-6 uger. Hæmoglobinresponset varierer mellem patienterne og kan påvirkes af jernaflejringer og tilstedeværelsen af samtidige medicinske problemer.
Kemoterapi induceret anæmi
Epoetin alfa administreret 3 gange om ugen eller en gang om ugen har vist sig at øge hæmoglobin og reducere behovet for transfusioner efter den første behandlingsmåned hos anæmiske kræftpatienter, der får kemoterapi.
I en undersøgelse, der sammenlignede doseringsregimer på 150 IE / kg 3 gange om ugen og 40.000 IE en gang om ugen hos raske forsøgspersoner og hos anæmiske kræftpersoner, var de tidsmæssige profiler af procentvise ændringer i reticulocytter, hæmoglobin og totale røde blodlegemer ens mellem de to doseringsregimer hos raske og anæmiske kræftpersoner. AUC'erne for de respektive farmakodynamiske parametre var ens mellem 150 IE / kg 3 gange om ugen og 40.000 IU doseringsregimer en gang ugentligt hos både raske og anæmiske kræftpersoner.
Voksne kirurgiske patienter i et autologt predonationsprogram
Epoetin alfa har vist sig at stimulere produktionen af røde blodlegemer for at øge autolog blodindsamling og for at begrænse faldet i hæmoglobin hos voksne patienter, der er planlagt til større elektiv kirurgi, for hvem der ikke forventes fuld perioperativ foropbevaring. Deres blodbehov. De største effekter blev set hos patienter med lave hæmoglobinniveauer (≤ 13 g / dl).
Behandling af voksne patienter planlagt til større elektiv ortopædkirurgi
Hos patienter, der er planlagt til større elektiv ortopædkirurgi med forbehandling af hæmoglobin> 10 til ≤ 13 g / dL, har epoetin alfa vist sig at reducere risikoen for at modtage allogene transfusioner og fremskynde genopretning af erythroid (øget hæmoglobinniveau, hæmatokrit og retikulocyttal).
Klinisk effekt og sikkerhed
Kronisk nyresvigt
Epoetin alfa er blevet evalueret i kliniske forsøg med anæmiske voksne patienter med CRF, herunder hæmodialyse og præ-dialyse patienter, til behandling af anæmi og opretholdelse af hæmatokrit inden for et koncentrationsinterval på 30 til 36%.
I kliniske forsøg med startdoser på 50-150 IE / kg tre gange om ugen reagerede cirka 95% af alle patienter med en klinisk signifikant stigning i hæmatokrit Efter ca. to måneders behandling blev næsten alle patienter transfunderet. -Uafhængig. Gang hæmatokritmålet blev nået, blev vedligeholdelsesdosis tilpasset til hver patient.
I de tre store kliniske forsøg med voksne dialysepatienter var den gennemsnitlige vedligeholdelsesdosis, der var nødvendig for at opretholde hæmatokrit mellem 30 og 36%, cirka 75 IE / kg 3 gange om ugen.
I et dobbeltblindet, multicenter, placebokontrolleret studie af livskvalitet for CRF-patienter ved hæmodialyse blev der påvist en klinisk og statistisk signifikant forbedring hos patienter behandlet med epoetin alfa sammenlignet med placebogruppen, der måler træthed, fysiske symptomer, forhold og depression (nyresygdomsspørgeskema) efter seks måneders behandling. Patienter i epoetin alfa-gruppen blev indskrevet i et åbent forlængelsesstudie, som viste, at forbedringer i deres livskvalitet blev opretholdt i yderligere 12 måneder.
Voksne patienter med nyreinsufficiens endnu ikke i dialyse
I kliniske undersøgelser af CRF -patienter, der ikke var i dialysebehandlet med epoetin alfa, var den gennemsnitlige varighed af behandlingen næsten fem måneder. Disse patienter reagerede på epoetin alfa -behandling på samme måde som hos dialysepatienter. CRF -patienter, der ikke var i dialyse, viste dosisafhængighed og konstant stigning i hæmatokrit, når epoetin alfa blev administreret både intravenøst og subkutant. Lignende hæmatokritvæksthastigheder blev noteret, da epoetin alfa blev administreret til begge. Desuden doser af epoetin alfa 75 til 150 IE / kg om ugen har vist sig at opretholde hæmatokrit på 36 til 38% i op til seks måneder.
I 2 forlængede doseringsintervalundersøgelser af EPREX (3 gange om ugen, en gang om ugen, en gang hver anden uge og en gang hver fjerde uge) opretholdt nogle patienter med længere doseringsintervaller ikke tilstrækkelige hæmoglobinniveauer og opfyldte de fastsatte abstinenskriterier (0% en gang ugentligt, 3,7% en gang hver anden uge og 3,3% i grupperne hver 4. uge).
En prospektiv randomiseret undersøgelse (CHOIR) vurderede 1432 anæmiske patienter med kronisk nyresvigt, der ikke undergik dialyse. Patienterne blev tildelt behandling med epoetin alfa med det formål at opretholde hæmoglobinniveau på 13,5 g / dl (over det anbefalede hæmoglobinniveau) eller 11,3 g / dl. En større kardiovaskulær hændelse (død, myokardieinfarkt, slagtilfælde eller hospitalsindlæggelse ved kongestivt hjertesvigt) , forekom blandt 125 (18%) af 715 patienter i den højere hæmoglobingruppe sammenlignet med 97 (14%) blandt 717 patienter i den lavere hæmoglobingruppe (procentdel af risiko [HR] 1,3, 95%CI: 1,0, 1,7, p = 0,03).
Kumulative retrospektive analyser af kliniske undersøgelser af ESA'er blev udført hos patienter med kronisk nyresvigt (hos dialyse- og ikke-dialysepatienter, diabetiske og ikke-diabetiske patienter). En tendens til øget estimeret risiko for alle årsager til dødelighed, kardiovaskulære og cerebrovaskulære hændelser forbundet med højere kumulative doser af ESA blev observeret uanset patientens diabetes eller dialysestatus (se pkt. 4.2 og pkt. 4.4).
Behandling af patienter med kemoterapi induceret anæmi
Epoetin alfa er blevet evalueret i kliniske forsøg med voksne anæmiske kræftpatienter med lymfoide og solide tumorer og hos patienter på forskellige kemoterapiregimer, herunder platin og ikke-platinholdige regimer. I disse undersøgelser viste epoetin alfa administreret 3 gange om ugen og en gang om ugen at øge hæmoglobin og reducere behovet for transfusioner efter den første behandlingsmåned hos anæmiske kræftpatienter. I nogle undersøgelser blev den dobbeltblindede fase efterfulgt af en åben fase, hvor alle patienter modtog epoetin alfa, og der blev observeret effekt.
Tilgængelige data tyder på, at patienter med hæmatologiske maligniteter og solide tumorer reagerer ækvivalent på epoetin alfa -behandling, og at patienter med eller uden tumorinfiltration af knoglemarven reagerer ækvivalent på epoetin alfa -behandling. Den sammenlignelige intensitet af kemoterapi i epoetin alfa- og placebogrupperne i kemoterapistudierne blev påvist af et lignende område under neutrofiltidskurven hos patienter behandlet med epoetin alfa og placebo samt af en lignende andel af patienterne i de behandlede grupper. med epoetin alfa og placebobehandlede grupper, hvis absolutte neutrofiltal faldt til under 1.000 og 500 celler / mcL
I en prospektiv, dobbeltblind, randomiseret og placebokontrolleret undersøgelse af 375 anæmiske patienter med ikke-myeloide tumorer, der modtager ikke-platinbaseret kemoterapi, en signifikant reduktion i anæmi-relaterede symptomer (såsom asteni, træthed og reduceret blodtryk) blev fundet. aktivitet), målt med følgende evalueringsskalaer: Funktionel vurdering af kræftterapi-anæmi (FACT-An) generel skala; FACT-En træthedsskala og Cancer Linear Analog Scale (CLAS).
To andre randomiserede og placebokontrollerede forsøg udført på et mindre antal patienter viste ikke nogen forbedring af livskvalitetsparametre ifølge EORTC-QLQ-C30- og CLAS-skalaerne.
Overlevelse og tumorprogression blev undersøgt i fem store kontrollerede undersøgelser, der omfattede i alt 2.833 patienter, heraf fire dobbeltblinde placebokontrollerede og et åbent mærke. Undersøgelserne indskrev patienter i både kemoterapibehandling (to undersøgelser) og patienter, hvor brugen af ESA'er ikke var indiceret: anæmiske kræftpatienter, der ikke modtog kemoterapi, og patienter med hoved- og halskræft, der modtog strålebehandling. Det ønskede koncentrationsniveau for strålebehandling. Hæmoglobin i to undersøgelser var> 13 g / dl; i de resterende tre undersøgelser var det 12 til 14 g / dl. I det åbne studie var der ingen forskel i overlevelse mellem patienter behandlet med rekombinant humant erythropoietin og kontroller. I de fire kontrollerede undersøgelser er"fareforhold for den samlede overlevelse varierede den fra 1,25 til 2,47 til fordel for kontroller. Disse undersøgelser viste en uforklarlig statistisk signifikant stigning i dødeligheden hos kræftanæmiske patienter behandlet med rekombinant humant erythropoietin sammenlignet med kontroller.Det samlede overlevelsesresultat for patienter behandlet med rekombinant humant erythropoietin kontra kontrol kunne ikke tilfredsstillende forklares med forskellen i forekomsten af trombose og relaterede komplikationer.
En patientniveauanalyse blev også udført på over 13.900 kræftpatienter (kemo-, radio-, radiokemio- eller ikke på terapi), som deltog i 53 kontrollerede kliniske forsøg med forskellige epoetiner. Metaanalysen af samlede overlevelsesdata indikerede et estimat af punktligt fareforhold på 1,06 til fordel for kontroller (95% CI: 1,00, 1,12; 53 undersøgelser og 13933 patienter), mens for kræftpatienter, der modtog kemoterapi, samlet overlevelse, mht. hazard ratio, var det 1,04 (95% CI: 0,97, 1,11; 38 undersøgelser og 10441 patienter). Endvidere indikerer metaanalyser konsekvent en signifikant øget relativ risiko for tromboemboliske hændelser hos kræftpatienter, der modtager rekombinante humane erythropoietiner (se pkt. 4.4).
Autologt predonationsprogram
Effekten af epoetin alfa til at lette autolog bloddonation hos patienter med lav hæmatokrit (≤ 39% og uden tydelig anæmi på grund af jernmangel), der var planlagt til større ortopædkirurgi, blev evalueret i en dobbelt placebokontrolleret undersøgelse. Blindet hos 204 patienter og en enkelt -blind placebokontrolleret undersøgelse hos 55 patienter.
I det dobbeltblinde studie blev patienter behandlet med epoetin alfa 600 IE / kg eller placebo intravenøst en gang dagligt hver 3. eller 4. dag i 3 uger (i alt 6 doser). I gennemsnit var patienter behandlet med epoetin alfa i stand til på forhånd at deponere betydeligt flere blodenheder (4,5 enheder) end patienter behandlet med placebo (3,0 enheder).
I det enkeltblinde studie blev patienter behandlet med epoetin alfa 300 IE / kg eller 600 IE / kg eller placebo intravenøst en gang dagligt hver 3. eller 4. dag i 3 uger (i alt 6 doser). Patienter behandlet med epoetin alfa var også i stand til på forhånd at deponere signifikante yderligere enheder blod (epoetin alfa 300 IE / kg = 4,4 enheder; epoetin alfa 600 IE / kg = 4,7 enheder) sammenlignet med patienter behandlet med placebo (2,9 enheder).
Epoetin alfa -behandling reducerede risikoen for allogen blodeksponering med 50% sammenlignet med patienter, der ikke blev behandlet med epoetin alfa.
Stor elektiv ortopædkirurgi
Effekten af epoetin alfa (300 IE / kg eller 100 IE / kg) på eksponering for allogene blodtransfusioner blev evalueret i et dobbeltblindet, placebokontrolleret klinisk studie hos jernmangel voksne patienter, der afventer operation. Elektiv ortopedisk kirurgi eller knæoperation Epoetin alfa blev administreret subkutant i 10 dage før operationen, på operationsdagen og i fire dage efter operationen. Patienter blev klassificeret i henhold til deres baseline hæmoglobin (≤ 10 g / dL,> 10 til ≤ 13 g / dL og> 13 g / dL).
Epoetin alfa 300 IE / kg reducerede signifikant risikoen for allogene transfusioner hos patienter med hæmoglobin før behandling fra> 10 til ≤ 13g / dL. 16% af patienterne behandlet med epoetin alfa 300 IE / kg, 23% med epoetin alfa 100 IE / kg og 45% med placebo krævede transfusion.
Et åbent parallelt gruppestudie med jernmangel hos voksne personer med hæmoglobin på forbehandling ≥ 10 til ≤ 13 g / dL, der ventede på ortopædisk hofte- eller knæoperation, sammenlignede behandling med epoetin alfa 300 IE / kg subkutant dagligt i 10 dage før operationen, den operationsdagen og i fire dage efter operationen med behandling med epoetin alfa 600 IE / kg subkutant en gang om ugen i 3 uger før operationen og på operationsdagen.
Fra forbehandling til præoperation var den gennemsnitlige stigning i hæmoglobin i den ugentlige 600 IE / kg (1,44 g / dl) gruppe det dobbelte af den observerede i den 300 IE / kg daglige gruppe (0, 73 g / dl). Gennemsnitlige hæmoglobinniveauer var ens i de to behandlingsgrupper i hele den postoperative periode.
Den erytropoietiske respons observeret i begge behandlingsgrupper førte til lignende transfusionshastigheder (16% i gruppen 600 IE / kg / uge og 20% i gruppen på 300 IE / kg / dag)
Pædiatrisk population
Kronisk nyresvigt
Epoetin alfa blev evalueret i et åbent, ikke-randomiseret, åbent dosisinterval, 52-ugers klinisk undersøgelse hos pædiatriske CRF-patienter, der gennemgik hæmodialyse. Gennemsnitsalderen for de patienter, der var indskrevet i undersøgelsen, var 11,6 år (interval 0,5 til - 20,1 år).
Epoetin alfa blev administreret med 75 IE / kg / uge intravenøst i 2 eller 3 opdelte doser efter dialyse, titreret fra 75 IE / kg / uge med 4 ugers mellemrum (op til maksimalt 300 IE / kg / uge.), for at opnå en stigning på 1 g / dl / måned i hæmoglobin Det ønskede hæmoglobinniveau var 9,6 til 11,2 g / dl. 81 procent af patienterne nåede målhæmoglobinniveauet. Mediantid til destination var 11 uger, median dosis til destination var 150 IE / kg / uge. Af de patienter, der nåede målet, gjorde 90% det på et doseringsregime 3 gange om ugen.
Efter 52 uger forblev 57% af patienterne i undersøgelsen og modtog en gennemsnitlig dosis på 200 IE / kg / uge.
05.2 Farmakokinetiske egenskaber
Absorption
Efter subkutan administration når serumniveauerne af epoetin alfa mellem 12 og 18 timer efter administration. Der var ingen ophobning efter administration af flere ugentlige doser på 600 IE / kg administreret subkutant.
Den absolutte biotilgængelighed af subkutant injiceret epoetin alfa er ca. 20% hos raske personer.
Fordeling
Den gennemsnitlige fordelingsvolumen er 49,3 ml / kg efter intravenøs administration af 50 og 100 IE / kg hos raske forsøgspersoner. Efter intravenøs administration af epoetin alfa til personer med kronisk nyresvigt varierede fordelingsvolumen fra 57-107 ml / kg efter enkelt administration (12 IE / kg) til 42-64 ml / kg efter administration af flere doser (48-192 IE / kg). Følgelig er fordelingsvolumen lidt større end plasmavolumen.
Eliminering
Halveringstiden for epoetin alfa efter intravenøs administration af flere doser er ca. 4 timer hos raske forsøgspersoner. Halveringstiden efter subkutan administration er estimeret til ca. 24 timer hos raske personer.
Den gennemsnitlige CL / F -værdi for 150 IE / kg 3 gange om ugen og 40.000 IE én gang ugentligt hos raske forsøgspersoner var henholdsvis 31,2 og 12,6 ml / t / kg. Den gennemsnitlige CL / F -værdi for 150 IE / kg 3 gange om ugen og 40.000 IE én gang ugentligt hos anemiske kræftpersoner var henholdsvis 45,8 og 11,3 ml / t / kg. Hos de fleste anæmiske kræftpersoner, der modtog cyklisk kemoterapi, var CL / F lavere efter subkutane doser på 40.000 IE en gang om ugen og 150 IE / kg 3 gange om ugen sammenlignet med værdierne for raske forsøgspersoner.
Linearitet / ikke-linearitet
Hos raske forsøgspersoner blev en dosisproportional stigning i serumkoncentrationer af epoetin alfa observeret efter intravenøs administration af 150 og 300 IE / kg, 3 gange om ugen. Enkelt subkutane doser på 300-2400 IE / kg epoetin alfa resulterede i et lineært forhold mellem middel C og dosis og mellem middel AUC og dosis. Der blev observeret et omvendt forhold mellem klareringtilsyneladende og dosis hos raske personer.
I undersøgelser, der undersøgte forlængelsen af intervallet mellem doser (40.000 IE en gang om ugen og 80.000, 100.000 og 120.000 IE hver uge), blev der observeret en lineær, men ikke-dosis proportional relation mellem middel Cmax og dosis el "Middel AUC og dosis ved steady state .
Farmakokinetisk / farmakodynamisk relation
Epoetin alfa udviser en dosisrelateret effekt på hæmatologiske parametre, som er uafhængig af indgivelsesvejen.
Pædiatrisk population
Der er rapporteret om en halveringstid på cirka 6,2-8,7 timer efter multipel intravenøs administration af epoetin alfa hos pædiatriske patienter med kronisk nyresvigt. Den farmakokinetiske profil af epoetin alfa hos børn og unge ser ud til at ligne den hos voksne.
Nyresvigt
Hos patienter med kronisk nyresvigt er halveringstiden for epoetin alfa, administreret intravenøst, lidt længere, cirka 5 timer, sammenlignet med raske forsøgspersoner.
05.3 Prækliniske sikkerhedsdata
I toksikologiske studier med gentagne doser hos hunde og rotter, men ikke hos aber, var epoetin alfa -behandling forbundet med en subklinisk tilstand af knoglemarvsfibrose. Knoglemarvsfibrose er en kendt komplikation af kronisk nyresvigt hos mennesker og kan være relateret til sekundær hyperparathyroidisme eller være forårsaget af ukendte faktorer.I en undersøgelse af hæmodialysepatienter behandlet med epoetin alfa i 3 år var forekomsten af knoglemarvsfibrose ikke højere end i gruppen af kontroldialysepatienter, der ikke blev behandlet med epoetin alfa.
Epoetin alfa fremkalder ikke genmutationer i bakterier (Ames -test), kromosomafvigelse i pattedyrceller, mikrokerner i mus eller genmutation på HGPRT -locus.
Langsigtede kræftfremkaldende undersøgelser er ikke blevet udført. Modstridende resultater i litteraturen baseret på dataene in vitro fra humane tumorprøver tyder på, at erythropoietiner kan spille en rolle i tumorproliferation.Dette er af usikker betydning i klinisk praksis.
I cellekulturer af humane knoglemarvsceller stimulerer epoetin alfa specifikt erythropoiesis og involverer ikke leukopoiesis. Ingen cytotoksisk aktivitet af epoetin alfa er blevet observeret på knoglemarvsceller.
I dyreforsøg har epoetin alfa vist sig at reducere fostrets kropsvægt, forsinke ossifikation og øge fosterdødeligheden, når den administreres i ugentlige doser cirka 20 gange den anbefalede ugentlige dosis til mennesker. Disse ændringer betragtes som sekundære til faldet i moderens kropsvægt, og betydningen for mennesker er ukendt ved terapeutiske doser.
06.0 LÆGEMIDDELOPLYSNINGER
06.1 Hjælpestoffer
Polysorbat 80
Glycin
Vand til injektionsvæsker.
Natriummonobasisk fosfatdihydrat
Dibasisk natriumphosphatdihydrat
Natriumchlorid
06.2 Uforenelighed
I mangel af kompatibilitetsundersøgelser må lægemidlet ikke blandes med andre produkter.
06.3 Gyldighedsperiode
18 måneder.
06.4 Særlige opbevaringsforhold
Opbevares i køleskab (2 ° C - 8 ° C). Dette temperaturområde skal sikres indtil tidspunktet for administration til patienten.
Opbevares i den originale beholder for at beskytte produktet mod lys.
Må ikke fryses eller rystes.
Til ambulant brug kan medicinen tages ud af køleskabet uden at blive udskiftet i en periode på maksimalt 3 dage ved en temperatur på højst 25 ° C. Hvis medicinen ikke er blevet brugt i slutningen af denne periode, skal den bortskaffes.
06.5 Den umiddelbare emballages art og emballagens indhold
0,3 ml (3.000 IE) injektionsvæske, opløsning i en fyldt injektionssprøjte (type I-glas) med stemplet (teflonbelagt gummi) og nål med hus (gummi med polypropylenbelægning) og PROTECS nålesikringsanordning (polycarbonat) fastgjort til sprøjten - pakke med 1.
0,4 ml (4.000 IE) i en fyldt injektionssprøjte (type I-glas) med stempel (teflonbelagt gummi) og nål med hus (gummi med polypropylenbelægning) og PROTECS nålesikringsanordning (polycarbonat) fastgjort til sprøjten-pakning med 1.
0,5 ml (5.000 IE) injektionsvæske, opløsning i en fyldt injektionssprøjte (type I-glas) med stemplet (teflonbelagt gummi) og nål med hus (gummi med polypropylenbelægning) og PROTECS nålesikringsanordning (polycarbonat) fastgjort til sprøjten - pakke med 1.
0,6 ml (6.000 IE) injektionsvæske, opløsning i en fyldt injektionssprøjte (type I-glas) med stempel (teflonbelagt gummi) og nål med hus (gummi med polypropylenbelægning) og PROTECS nålesikringsanordning (polycarbonat) fastgjort til sprøjten - pakke med 1.
0,8 ml (7.000 IE) injektionsvæske, opløsning i en fyldt injektionssprøjte (type I-glas) med stempel (teflonbelagt gummi) og nål med hus (gummi med polypropylenbelægning) og PROTECS nålesikringsanordning (polycarbonat) fastgjort til sprøjten - pakke med 1.
1,0 ml (10.000 IE) injektionsvæske, opløsning i en fyldt injektionssprøjte (type I-glas) med stemplet (teflonbelagt gummi) og nål med hus (gummi med polypropylenbelægning) og PROTECS nålesikringsanordning (polycarbonat) fastgjort til sprøjten - pakke med 1.
06.6 Brugsanvisning og håndtering
Produktet må ikke bruges og skal smides:
• Hvis forseglingen er brudt;
• Hvis opløsningen er farvet eller i nærvær af partikler;
• Hvis der er opstået indefrysning eller er mistænkt;
• Hvis der er fejl i køleskabet.
Engangsprodukt. Administrer ikke mere end én dosis pr. Sprøjte efter fjernelse af den uønskede mængde opløsning fra sprøjten. Se afsnit 3. Sådan bruges EPREX (injektionsinstruktioner) i indlægssedlen.
De fyldte sprøjter er udstyret med en PROTECS nålesikring for at forhindre risiko for nålestik. Indlægssedlen indeholder alle oplysninger om brug og håndtering af de fyldte sprøjter med sikkerhedsanordningen. PROTECS nål.
Ubrugt medicin og affald fra denne medicin skal bortskaffes i overensstemmelse med lokale regler.
07.0 INDEHAVER AF MARKEDSFØRINGSTILLADELSE
Janssen -Cilag SpA, via M.Buonarroti, 23. - 20093 Cologno Monzese (MI)
08.0 MARKEDSFØRINGSTILLADELSESNUMMER
027015167 - "10.000 IE / ML injektionsvæske, opløsning i fyldt injektionssprøjte" 1 sprøjte på 3.000 IE / 0.3ML
027015179 - "10.000 IE / ML injektionsvæske, opløsning i fyldt injektionssprøjte" 1 sprøjte med 4.000 IE / 0.4ML
027015231 - "10.000 IE / ML injektionsvæske, opløsning i fyldt injektionssprøjte" 1 sprøjte med 5.000 IE / 0.5ML
027015243 - "10.000 IE / ML injektionsvæske, opløsning i fyldt injektionssprøjte" 1 sprøjte med 6.000 IE / 0.6ML
027015268 - "10.000 IE / ML injektionsvæske, opløsning i fyldt injektionssprøjte" 1 sprøjte med 8.000 IE / 0,8 ml
027015181 - "10.000 IE / ML injektionsvæske, opløsning i fyldt injektionssprøjte" 1 sprøjte med 10.000 IE / 1ML
09.0 DATO FOR FØRSTE TILLADELSE ELLER FORNYELSE AF TILLADELSEN
Maj 1989
AIC -fornyelse: 4. august 2008
10.0 DATO FOR REVISION AF TEKSTEN
08/2015