Aktive ingredienser: Sorafenib
Nexavar 200 mg filmovertrukne tabletter
Hvorfor bruges Nexavar? Hvad er det for?
Nexavar bruges til behandling af hepatocarcinom.
Nexavar bruges også til behandling af nyrekræft (fremskreden nyrecellecarcinom), når det er på et fremskredent stadium, og når standardterapi ikke har hjulpet med at stoppe det eller anses for uegnet.
Nexavar bruges til behandling af kræft i skjoldbruskkirtlen (differentieret kræft i skjoldbruskkirtlen).
Nexavar er en såkaldt multikinasehæmmer. Det virker ved at bremse væksthastigheden af kræftceller og blokere blodtilførslen, der gør det muligt for kræftceller at vokse.
Kontraindikationer Når Nexavar ikke bør anvendes
Tag ikke Nexavar
- hvis du er allergisk over for sorafenib eller et af de øvrige indholdsstoffer i dette lægemiddel (angivet i afsnit 6).
Forholdsregler ved brug Det, du skal vide, før du tager Nexavar
Tal med din læge eller apotek, før du tager Nexavar.
Vær særlig forsigtig med Nexavar
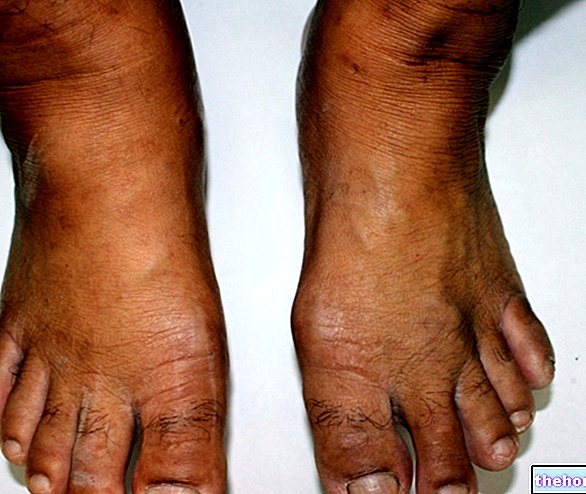
- Hvis der opstår hudproblemer. Nexavar kan forårsage udslæt og hudreaktioner, især på hænder og fødder. Disse virkninger kan normalt behandles af lægen. Ellers kan lægen afbryde behandlingen eller stoppe den helt.
- Hvis du har forhøjet blodtryk. Nexavar kan forårsage en stigning i blodtrykket; din læge vil kontrollere dit blodtryk regelmæssigt og kan ordinere medicin til behandling af forhøjet blodtryk.
- Hvis du har blødningsproblemer, eller hvis du tager warfarin eller phenprocomon. Behandling med Nexavar kan føre til en øget risiko for blødning. Hvis du tager warfarin eller phenprocomon, medicin, der fortynder blodet for at forhindre blodpropper, kan der være en øget risiko for blødning.
- Hvis du har smerter i brystet eller hjerteproblemer. Din læge kan beslutte at stoppe behandlingen eller stoppe den helt.
- Hvis du har en hjertelidelse, f.eks. En 'elektrisk signalforstyrrelse kaldet' QT -forlængelse '.
- Hvis du er ved at blive opereret eller lige er blevet opereret. Nexavar kan påvirke sårheling. Hvis du er ved at blive opereret, vil din behandling med Nexavar sandsynligvis blive stoppet. Din læge vil derefter beslutte, hvornår den skal tages tilbage.
- Hvis du behandles med irinotecan eller docetaxel, som også er medicin mod kræft, kan Nexavar øge virkningerne og især bivirkningerne af disse lægemidler.
- Hvis du tager neomycin eller andre antibiotika. Nexavars effektivitet kan være nedsat - hvis du har alvorligt leversvigt Du kan få forværrede bivirkninger, når du tager denne medicin.
- Hvis du har nedsat nyrefunktion. Din læge vil overvåge din vand- og elektrolytbalance.
- Fertilitet. Nexavar kan reducere fertiliteten hos både mænd og kvinder. Tal med din læge, hvis dette gælder for dig.
- Gastrointestinal perforering kan forekomme under behandlingen (se afsnit 4: Mulige bivirkninger). I dette tilfælde vil lægen stoppe behandlingen.
- Hvis du har kræft i skjoldbruskkirtlen, vil din læge kontrollere niveauerne af calcium og skjoldbruskkirtelhormon i dit blod.
Fortæl det til din læge, hvis noget af dette gælder for dig. Du har muligvis brug for behandling af disse problemer, eller din læge kan ændre dosis af Nexavar eller stoppe behandlingen helt (se også afsnit 4: Mulige bivirkninger).
Børn og unge
Nexavar er endnu ikke undersøgt hos børn og unge.
Interaktioner Hvilke lægemidler eller fødevarer kan ændre virkningen af Nexavar
Nogle lægemidler kan påvirke Nexavar eller blive påvirket af det. Fortæl det til din læge eller apotek, hvis du tager, for nylig har taget eller måske tager nogen af lægemidlerne på denne liste eller andre lægemidler, herunder receptpligtige:
- Rifampicin, neomycin eller andre lægemidler til behandling af infektioner (antibiotika)
- Hypericum perforatum, også kendt som "perikon", en urtebehandling mod depression
- Phenytoin, carbamazepin eller phenobarbital, behandlinger mod epilepsi og andre sygdomme
- Dexamethason, et kortikosteroid, der bruges til forskellige sygdomme
- Warfarin eller phenprocomon, antikoagulantia, der bruges til at forhindre blodpropper
- Doxorubicin, capecitabin, docetaxel, paclitaxel og irinotecan, der bruges til behandling af kræft.
- Digoxin, der bruges til behandling af let eller moderat hjertesvigt
Advarsler Det er vigtigt at vide, at:
Graviditet og amning
Undgå at blive gravid, mens du bliver behandlet med Nexavar. Hvis du er i den fertile alder, skal du bruge effektiv prævention under behandling med Nexavar. Hvis du bliver gravid, mens du bliver behandlet med Nexavar, skal du straks fortælle det til din læge, hvem der afgør, om behandlingen skal fortsættes eller stoppes.
Du bør ikke amme din baby, mens du bliver behandlet med Nexavar, da denne medicin kan forstyrre din babys vækst og udvikling.
Kørsel og brug af maskiner
Der er ingen grund til at tro, at Nexavar påvirker evnen til at føre motorkøretøj eller betjene maskiner.
Dosis, metode og administrationstidspunkt Sådan bruges Nexavar: Dosering
Den anbefalede dosis Nexavar til voksne er to 200 mg tabletter to gange dagligt.
Disse svarer til en daglig dosis på 800 mg eller fire tabletter om dagen. Tag Nexavar tabletter med et glas vand, mellem måltiderne eller med fødevarer med lav til mellem fedt. Tag ikke denne medicin med meget fede fødevarer, da disse kan reducere deres effektivitet. Hvis du planlægger at spise meget fed mad, skal du tage tabletterne mindst 1 time før eller 2 timer efter frokost. Tag altid denne medicin nøjagtigt efter lægens anvisning. Kontakt din læge eller apotek, hvis du er i tvivl.
Det er vigtigt at tage denne medicin på omtrent samme tid hver dag for at holde koncentrationen i blodet konstant.
Denne medicin tages normalt, så længe der er noteret kliniske fordele, og der er ingen utålelige bivirkninger.
Overdosering Hvad skal jeg gøre, hvis du har taget for meget Nexavar
Hvis du har taget for mange Nexavar
Fortæl det straks til din læge, hvis du eller nogen anden har taget mere end den foreskrevne dosis. At tage for meget Nexavar gør bivirkninger mere sandsynlige eller mere alvorlige, især diarré og hudreaktioner. Din læge kan fortælle dig, at du skal stoppe med at tage denne medicin.
Hvis du har glemt at tage Nexavar
Hvis du har glemt at tage en dosis, skal du tage den, så snart du husker det. Hvis din næste dosis er kort tid, skal du glemme den glemte dosis og fortsætte med din frekvens
Bivirkninger Hvad er bivirkningerne af Nexavar
Ligesom al anden medicin kan denne medicin forårsage bivirkninger, men ikke alle får bivirkninger. Denne medicin kan også ændre resultaterne af nogle blodprøver.
Meget normal:
kan påvirke mere end 1 ud af 10 personer
- diarré
- utilpashed (kvalme)
- føler sig svag eller træt (træthed)
- smerter (herunder smerter i munden, mave, hovedpine, knoglesmerter, kræftsmerter)
- hårtab (alopeci)
- rødme eller smerter i håndfladerne eller fodsålerne (hånd-fod hudreaktion)
- kløe eller udslæt
- Han trak sig tilbage
- blødning (herunder blødning i hjernen, tarmvæggen og luftvejene)
- forhøjet blodtryk eller forhøjet blodtryk (hypertension)
- infektioner
- appetitløshed (anoreksi)
- forstoppelse
- ledsmerter (artralgi)
- feber
- vægttab
- tørhed i huden
Almindelige:
kan ramme op til 1 ud af 10 personer
- influenzalignende sygdom
- fordøjelsesbesvær (dyspepsi)
- synkebesvær (dysfagi)
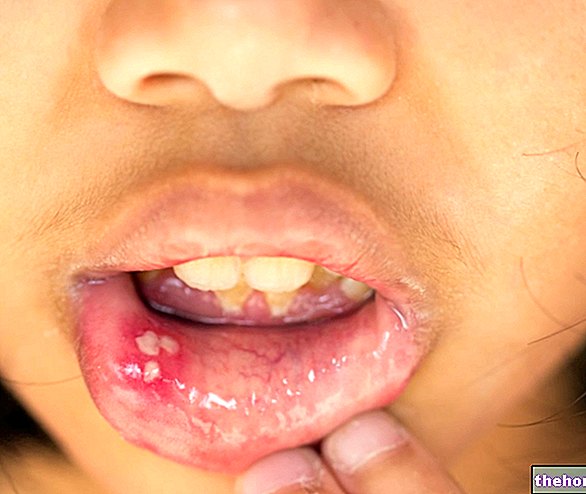
- betændelse eller mundtørhed, smerter i tungen (stomatitis og betændelse i slimhinden)
- lave niveauer af calcium i blodet (hypocalcæmi)
- lavt indhold af kalium i blodet (hypokaliæmi)
- muskelsmerter (myalgi)
- følsomhedsforstyrrelser i fingre og tæer, herunder prikken og følelsesløshed (perifer sensorisk neuropati)
- depression
- erektionsproblemer (impotens)
- stemmeændringer (dysfoni)
- acne
- betændt, tør eller flagnende hud (dermatitis, hudskalning)
- hjertefejl
- hjerteanfald (myokardieinfarkt) eller brystsmerter
- tinnitus (ringen i ørerne)
- nyresvigt
- høje niveauer af protein i urinen (proteinuri)
- generel svaghed eller tab af styrke (asteni)
- reduceret antal hvide blodlegemer (leukopeni og neutropeni)
- reduceret antal røde blodlegemer (anæmi)
- lavt antal blodplader i blodet (trombocytopeni)
- betændelse i hårsækkene (folliculitis)
- nedsat skjoldbruskkirtelaktivitet (hypothyroidisme)
- lavt natriumindhold i blodet (hyponatriæmi)
- ændringer i smagssansen (dysgeusi)
- rødme i ansigtet og ofte andre områder af huden (rødme)
- løbende næse (løbende næse)
- halsbrand (gastroøsofageal reflukssygdom)
- hudkræft (keratoacanthoma / pladecellecellekræft)
- fortykkelse af det ydre lag af huden (hyperkeratose)
- pludselig ufrivillig sammentrækning af en muskel (muskelspasmer)
Ualmindelig:
kan ramme op til 1 ud af 100 mennesker
- mavebetændelse (gastritis)
- mave (mave) smerter på grund af pancreatitis, betændelse i galdeblæren og / eller galdegangene
- gulfarvning af hud eller øjne (gulsot) forårsaget af høje niveauer af galdepigmenter (hyperbilirubinæmi)
- allergiske reaktioner (herunder hudreaktioner og nældefeber)
- dehydrering
- brystforstørrelse (gynækomasti)
- åndedrætsbesvær (lungesygdom)
- eksem
- overdreven skjoldbruskkirtelaktivitet (hypertyreose)
- flere hududslæt (erythema multiforme)
- højt blodtryk
- gastrointestinal perforering
- reversibelt ødem i bagsiden af hjernen, som kan være forbundet med hovedpine, ændret bevidsthed, anfald og visuelle symptomer, herunder synstab (posterior reversibel leukoencephalopati)
- pludselig, alvorlig allergisk reaktion (anafylaktisk reaktion)
Sjælden:
kan ramme op til 1 ud af 1.000 mennesker
- allergisk reaktion med hævelse af huden (f.eks. ansigt, tunge), som kan forårsage vejrtræknings- og synkebesvær (angioødem)
- unormal hjerterytme (QT -forlængelse)
- Betændelse i leveren, som kan føre til kvalme, opkastning, mavesmerter og gulsot (lægemiddelinduceret hepatitis)
- et "solskoldningslignende udslæt på huden, der tidligere har været udsat for strålebehandling og kan være alvorlig (aktinisk-lignende dermatitis)
- alvorlige hudreaktioner og / eller slimhinder, som kan omfatte smertefulde blærer og feber, med frigørelse af store hudområder (Stevens-Johnsons syndrom og toksisk epidermal nekrolyse)
- unormal muskelskade, som kan føre til nyreproblemer (rabdomyolyse)
- nyreskade, der får store mængder protein til at gå tabt i urinen (nefrotisk syndrom)
- betændelse i blodkarrene i huden, som kan vise sig som udslæt (leukocytoklastisk vaskulitis)
Ikke kendt:
hyppigheden kan ikke estimeres ud fra de tilgængelige data
- nedsat hjernefunktion, som kan være forbundet med f.eks. søvnighed, adfærdsændringer eller forvirring (encefalopati)
Indberetning af bivirkninger
Tal med din læge eller apotek, hvis du får bivirkninger, herunder mulige bivirkninger, som ikke er nævnt i denne indlægsseddel. Du kan også indberette bivirkninger direkte via det nationale rapporteringssystem anført i tillæg V. Ved at rapportere bivirkninger kan du hjælpe med at give mere information om sikkerheden ved dette lægemiddel.
Udløb og opbevaring
Opbevar denne medicin utilgængeligt for børn.
Brug ikke dette lægemiddel efter den udløbsdato, der står på kartonen efter EXP og på hver blister efter EXP. Udløbsdatoen refererer til den sidste dag i den pågældende måned.
Opbevar ikke dette lægemiddel over 25 ° C.
Smid ikke medicin via spildevand eller husholdningsaffald. Spørg din apotek om, hvordan du skal smide medicin, du ikke længere bruger. Dette vil hjælpe med at beskytte miljøet.
Nexavar indeholder
- Den aktive ingrediens er sorafenib. Hver filmovertrukket tablet indeholder 200 mg sorafenib (som tosylat).
- Øvrige indholdsstoffer er: Tabletkerne: croscarmellosenatrium, mikrokrystallinsk cellulose, hypromellose, natriumlaurylsulfat og magnesiumstearat. Tabletovertræk: hypromellose, macrogol, titandioxid (E 171) og rødt jernoxid (E 172)
Hvordan Nexavar ser ud og pakningens indhold
Nexavar 200 mg filmovertrukne tabletter er røde og runde, med Bayer-krydset på den ene side og "200" på den anden side. De findes i kartoner med 112 tabletter, der indeholder fire klare kalenderblister på 28 tabletter hver.
Indlægsseddel: AIFA (Italian Medicines Agency). Indhold offentliggjort i januar 2016. De foreliggende oplysninger er muligvis ikke opdaterede.
For at få adgang til den mest opdaterede version er det tilrådeligt at få adgang til webstedet AIFA (Italian Medicines Agency). Ansvarsfraskrivelse og nyttige oplysninger.
01.0 LÆGEMIDLETS NAVN
NEXAVAR 200 MG
02.0 KVALITATIV OG KVANTITATIV SAMMENSÆTNING
Hver filmovertrukket tablet indeholder 200 mg sorafenib (som tosylat).
Den fulde liste over hjælpestoffer findes i afsnit 6.1.
03.0 LÆGEMIDDELFORM
Filmovertrukket tablet (tablet).
Røde, runde, bikonvekse, filmovertrukne tabletter mærket med et Bayer-kryds på den ene side og "200" på den anden.
04.0 KLINISKE OPLYSNINGER
04.1 Terapeutiske indikationer
Hepatocarcinom
Nexavar er indiceret til behandling af hepatocellulært carcinom (se pkt.5.1).
Nyrecellekarcinom
Nexavar er indiceret til behandling af patienter med fremskreden nyrecellecarcinom, der tidligere har mislykkedes interferon alfa- eller interleukin-2-behandling, eller som anses for ikke at være berettiget til sådan behandling.
Differentieret kræft i skjoldbruskkirtlen
Nexavar er indiceret til behandling af patienter med lokalt fremskreden eller metastatisk, progressiv, radioiodion ildfast differentieret skjoldbruskkirtelkræft (papillær / follikulær / Hürthle -celle).
04.2 Dosering og indgivelsesmåde
Behandling med Nexavar bør være under opsyn af en læge med erfaring i brug af kræftbehandlinger.
Dosering
Den anbefalede dosis Nexavar til voksne er 400 mg sorafenib (to 200 mg tabletter) to gange dagligt (svarende til en samlet daglig dosis på 800 mg).
Behandlingen bør fortsætte, så længe der ses klinisk fordel, eller indtil der opstår uacceptabel toksicitet.
Justering af doseringen
Håndtering af formodede bivirkninger kan kræve midlertidig afbrydelse eller dosisreduktion af sorafenibbehandling.
Når en dosisreduktion er påkrævet under behandlingen af hepatocellulært carcinom (hepatocellulært carcinom, HCC) og nyrecellekarcinom (nyrecellekræft, RCC), bør dosis af Nexavar reduceres til to 200 mg sorafenib tabletter en gang dagligt (se pkt. 4.4).
Når en dosisreduktion er påkrævet under behandlingen af differentieret skjoldbruskkirtelkræft (differentieret thyreoideacancer, DTC), bør Nexavar -dosis reduceres til 600 mg sorafenib om dagen i opdelte doser (to 200 mg tabletter og en 200 mg tablet med tolv timers mellemrum).
Hvis yderligere dosisreduktion er påkrævet, kan Nexavar reduceres til 400 mg sorafenib om dagen i opdelte doser (to 200 mg tabletter med tolv timers mellemrum) og om nødvendigt reduceres yderligere til en 200 mg tablet én gang dagligt Efter forbedring af ikke-hæmatologisk bivirkninger, kan dosis af Nexavar øges.
Pædiatrisk population
Nexavars sikkerhed og virkning hos ældre børn og unge
Ældre befolkning
For den ældre befolkning (patienter over 65 år) er dosisjustering ikke nødvendig.
Nedsat nyrefunktion
Ingen dosisjustering er nødvendig for patienter med let, moderat eller svært nedsat nyrefunktion. Der er ingen data tilgængelige for patienter i dialyse (se pkt. 5.2).
Overvågning af vand og elektrolytbalance er tilrådeligt hos patienter med risiko for nyreinsufficiens.
Nedsat leverfunktion
Ingen dosisjustering er nødvendig for patienter med Child Pugh A eller B (let til moderat) nedsat leverfunktion. Der er ingen data tilgængelige om patienter med alvorligt nedsat leverfunktion ved Child Pugh C (se pkt. 4.4 og 5.2).
Indgivelsesmåde
Til oral brug
Sorafenib bør administreres mellem måltiderne eller et måltid med et lavt eller moderat fedtindhold. Hvis patienten har til hensigt at spise et måltid med højt fedtindhold, skal sorafenib tabletter tages mindst en time før eller to timer efter måltidet. Tabletterne skal synkes med et glas vand.
04.3 Kontraindikationer
Overfølsomhed over for det aktive stof eller over for et eller flere af hjælpestofferne anført i pkt.6.1.
04.4 Særlige advarsler og passende forholdsregler ved brug
Dermatologisk toksicitet
Hånd-fod hudreaktion (palmar-plantar erythrodysæstesi) e udslæt repræsenterer de mest almindelige bivirkninger af sorafenib. Udslæt og hånd-fod hudreaktion er normalt grad 1 og 2, ifølge i Fælles toksicitetskriterier (CTC), og forekommer generelt i løbet af de første seks uger af sorafenib -behandling. Håndtering af dermatologisk toksicitet kan omfatte topiske terapier til lindring af symptomer, midlertidig afbrydelse af behandlingen og / eller ændring af doseringen af sorafenib eller i alvorlige eller vedvarende tilfælde definitiv afbrydelse af administrationen (se pkt. 4.8).
Forhøjet blodtryk
En højere forekomst af arteriel hypertension blev observeret hos patienter behandlet med sorafenib. Hos disse patienter var hypertension normalt mild til moderat, forekom i de tidlige behandlingsstadier og reagerede på standard antihypertensiv behandling. Blodtrykket bør overvåges regelmæssigt og behandles efter behov i henhold til gældende medicinsk praksis. I tilfælde af alvorlig eller vedvarende hypertension eller hypertensiv krise, på trods af påbegyndelse af antihypertensiv behandling, anbefales det at overveje permanent sorafenib -administration afbrydes (se pkt. 4.8).
Blødning
Risikoen for blødning kan stige efter administration af sorafenib. Hvis en blødningsepisode kræver medicinsk indgreb, anbefales det at overveje permanent at afbryde administrationen af sorafenib (se pkt. 4.8).
Hjerteiskæmi og / eller hjerteanfald
I et dobbeltblindet, randomiseret, placebokontrolleret studie (studie 1, se afsnit 5.1) var forekomsten af behandlingsstartet hjerteinfarkt eller iskæmi højere i sorafenibgruppen (4,9%) end i behandlingsgruppen. Med placebo (0,4 I studie 3 (se afsnit 5.1) var forekomsten af behandlingsstartet hjerteinfarkt eller iskæmi 2,7% hos sorafenibbehandlede patienter og 1,3% hos patienter behandlet med placebo. Patienter med ustabil koronararteriesygdom eller med nylig myokardieinfarkt blev ekskluderet fra disse undersøgelser. Behovet for midlertidig eller permanent seponering af sorafenib -behandling bør overvejes hos patienter, der udvikler hjerteisæmi og / eller infarkt (se pkt. 4.8).
QT interval forlængelse
Sorafenib har vist sig at forlænge QT / QTc -intervallet (se afsnit 5.1), hvilket kan føre til en øget risiko for ventrikulær arytmi Brug sorafenib med forsigtighed hos patienter, der har eller kan udvikle QTc -forlængelse, f.eks. Patienter med en medfødt lang QT Syndrom, dem, der behandles med en høj kumulativ dosis af antracykliner, patienter, der tager visse antiarytmiske lægemidler eller andre lægemidler, der kan føre til forlængelse af QT, og dem med elektrolytforstyrrelser, for eksempel hypokaliæmi, hypocalcæmi eller hypomagnesæmi Når sorafenib anvendes til disse patienter, periodisk elektrokardiografi og elektrolyt (magnesium, kalium og calcium) målinger bør udføres i behandlingsperioden.
Gastrointestinal perforering
Gastrointestinal perforering er en usædvanlig hændelse og er blevet rapporteret hos mindre end 1% af patienterne, der tager sorafenib. I nogle tilfælde var der ingen sammenhæng med åbenbar intra-abdominal tumor. I tilfælde af gastrointestinal perforering bør administration af sorafenib seponeres (se pkt.4.8).
Nedsat leverfunktion
Der er ingen data tilgængelige om patienter med svært nedsat leverfunktion (Child Pugh C). Hos sådanne patienter kan eksponeringen øges, da sorafenib primært elimineres via leveren (se pkt. 4.2 og 5.2).
Samtidig administration af warfarin
Sjældne blødningsepisoder eller en stigning i INR (International Normaliseret
Forhold) er blevet rapporteret hos nogle patienter, der tog warfarin under behandling med sorafenib. Patienter i warfarin- eller phenprocoumon -behandling bør monitoreres regelmæssigt for ændringer i protrombintid, INR eller klinisk relevante blødningsepisoder (se pkt. 4.5 og 4.8).
Komplikationer ved sårheling
Der er ikke udført formelle undersøgelser af sorafenibs virkning på sårheling. Midlertidig suspension af sorafenib -behandling anbefales af forsigtighedshensyn hos patienter, der gennemgår større operationer. Klinisk erfaring med, hvornår behandling skal genstartes efter større operation er begrænset. Beslutningen om at genoptage behandling med sorafenib efter større operation bør derfor baseres på en klinisk vurdering af tilstrækkelig sårheling.
Ældre befolkning
Der er rapporteret tilfælde af nyresvigt. Derfor bør overvågning af nyrefunktionen overvejes.
Interaktion mellem lægemidler
Der udvises forsigtighed ved administration af sorafenib med stoffer, der hovedsageligt metaboliseres og / eller elimineres via UGT1A1 (f.eks. Irinotecan) eller UGT1A9 (se pkt. 4.5).
Forsigtighed anbefales ved samtidig administration af sorafenib og docetaxel (se pkt. 4.5).
Kombinationen med neomycin eller med andre antibiotika, der er i stand til at forårsage alvorlige økologiske forstyrrelser i den gastrointestinale mikroflora, kan føre til et fald i biotilgængeligheden af sorafenib (se pkt. 4.5) Risikoen for et fald i plasmakoncentrationen af sorafenib bør vurderes inden start. et behandlingsforløb med antibiotika.
Højere dødelighed blev observeret hos patienter med pladecellelungekræft behandlet med sorafenib i kombination med platinbaseret kemoterapi.
I to randomiserede kliniske forsøg, der undersøgte patienter med ikke-småcellet lungekræft (Ikke-småcellet lungekræft, NSCLC), hazard ratio (HR) for samlet overlevelse i en undergruppe af patienter med pladecellelungekræft var 1,81 (95% CI 1,19, 2,74) hos patienter behandlet med sorafenib ud over paclitaxel / carboplatin -behandling og 1,22 (95% CI 0,82; 1,80) hos patienter behandlet med sorafenib ud over gemcitabin / cisplatinbehandling. Der blev ikke observeret nogen dominerende dødsårsag, men en øget forekomst af respirationssvigt, blødning og infektion blev observeret hos patienter behandlet med sorafenib ud over platinbaseret terapi.
Patologiske specifikke advarsler
Differentieret skjoldbruskkirtelkræft (DTC)
Inden behandling påbegyndes, anbefales det, at læger omhyggeligt evaluerer individuel patientprognose baseret på maksimal læsionsstørrelse (se pkt.5.1), sygdomsrelaterede symptomer (se pkt.5.1) og progression.
Håndtering af formodede bivirkninger kan nødvendiggøre en "midlertidig afbrydelse eller dosisreduktion af sorafenib -terapi. I studie 5 (se pkt. 5.1) afbrød 37% af forsøgspersonerne behandlingen midlertidigt og 35% reducerede dosis allerede i cyklus 1 af sorafenib -behandling.
Dosisreduktion var kun delvist effektiv til at lindre bivirkninger. Gentagne fordele og risikovurderinger anbefales derfor under hensyntagen til antitumoraktivitet og tolerabilitet.
Blødning i DTC
På grund af den potentielle risiko for blødning skal tracheal, bronchial og esophageal infiltration behandles med lokal terapi, før sorafenib administreres til patienter med DTC.
Hypocalcæmi i DTC
Ved brug af sorafenib til patienter med DTC anbefales tæt overvågning af calciumniveauer i blodet. I kliniske forsøg var hypokalcæmi hyppigere og mere alvorlig hos patienter med DTC, især hos patienter med hypoparathyroidisme tidligere, sammenlignet med patienter med nyrecellecarcinom eller hepatocarcinom. Grad 3 og 4 hypokalcæmi forekom. Manifesteret hos 6,8% og 3,4% af sorafenib-behandlede patienter med DTC (se pkt. 4.8). Alvorlig hypokalcæmi bør korrigeres for at forhindre komplikationer såsom forlængelse af QT -interval eller torsades de pointes (se afsnittet forlængelse af QT -interval).
Undertrykkelse af TSH i DTC
I studie 5 (se pkt. 5.1) blev stigninger i TSH -niveauer større end 0,5 mU / L observeret hos patienter behandlet med sorafenib. Tæt overvågning af TSH -niveauer anbefales ved brug af sorafenib til patienter med DTC.
Nyrecellekarcinom
Højrisiko patienter, som defineret af MSKCC prognostiske gruppe (Memorial Sloan Kettering Cancer Center), blev ikke inkluderet i det kliniske fase III-studie med nyrecellekarcinom (se undersøgelse 1 i afsnit 5.1), og fordel-risiko-forholdet hos disse patienter er ikke blevet fastslået.
04.5 Interaktioner med andre lægemidler og andre former for interaktion
Fremkaldere af metaboliske enzymer
Administration af rifampicin i 5 dage før administration af en enkelt dosis sorafenib resulterede i en gennemsnitlig reduktion af sorafenib AUC på 37%. Andre inducere af CYP3A4 og / eller glucuronidering (f.eks. hypericum perforatum også kendt som "perikon", phenytoin, carbamazepin, phenobarbital og dexamethason) kan øge metabolismen af sorafenib og derved reducere dets koncentration.
CYP3A4 -hæmmere
Ketoconazol, en kraftig CYP3A4 -hæmmer, administreret en gang dagligt i 7 dage til raske mandlige frivillige, ændrede ikke den gennemsnitlige AUC for en enkelt dosis på 50 mg sorafenib Disse data tyder på, at de kliniske farmakokinetiske interaktioner af sorafenib med CYP3A4 -hæmmere er usandsynlige.
CYP2B6, CYP2C8 og CYP2C9 substrater
In vitro sorafenib hæmmer CYP2B6, CYP2C8 og CYP2C9 med næsten samme styrke. I kliniske farmakokinetiske undersøgelser resulterede co-administration af sorafenib 400 mg to gange dagligt med cyclophosphamid, et CYP2B6-substrat eller paclitaxel, et CYP2C8-substrat imidlertid ikke i klinisk signifikant hæmning. Disse data tyder på, at sorafenib ved den anbefalede dosis på 400 mg to gange dagligt, er muligvis ikke en hæmmer in vivo CYP2B6 eller CYP2C8.
Samtidig behandling med sorafenib og warfarin, et substrat for CYP2C9, førte ikke til ændringer i den gennemsnitlige PT-INR sammenlignet med placebo. Derfor også risikoen for en "hæmning in vivo klinisk relevant CYP2C9 af sorafenib kan betragtes som lav. Patienter, der tager warfarin eller phenprocoumon, bør imidlertid have deres INR overvåget regelmæssigt (se pkt. 4.4).
CYP3A4, CYP2D6 og CYP2C19 substrater
Samtidig administration af sorafenib og midazolam, dextromethorphan eller omeprazol, som er substrater for henholdsvis cytochromerne CYP3A4, CYP2D6 og CYP2C19, ændrede ikke eksponeringen for disse midler. Dette indikerer, at sorafenib hverken er en hæmmer eller en inducer af disse isoenzymer. cytokrom P450, derfor er kliniske farmakokinetiske interaktioner mellem sorafenib og substrater af disse enzymer usandsynlige.
UGT1A1 og UGT1A9 substrater
In vitro, sorafenib hæmmede glucuronidering ved UGT1A1 og UGT1A9. Den kliniske relevans af dette fund er ukendt (se nedenfor og pkt. 4.4).
Uddannelse in vitro om induktion af enzymer i CYP -systemet
CYP1A2 og CYP3A4 aktiviteter blev ikke ændret efter eksponering af humane hepatocytkulturer for sorafenib, hvilket indikerer, at sorafenib sandsynligvis ikke er en inducer af CYP1A2 og CYP3A4.
Substrater til P-gp
In vitro, sorafenib har vist sig at hæmme transportproteinet p-glycoprotein (P-gp). I tilfælde af samtidig behandling med sorafenib kan en stigning i plasmakoncentrationen af substrater for P-gp, såsom digoxin, ikke udelukkes.
Forening med andre antineoplastiske midler
I kliniske undersøgelser blev sorafenib administreret med en række andre antineoplastiske midler i deres almindeligt anvendte dosering, herunder gemcitabin, cisplatin, oxaliplatin, paclitaxel, carboplatin, capecitabin, doxorubicin, irinotecan, docetaxel og cyclophosphamid. Sorafenib havde ingen klinisk relevant effekt på gemcitabin, cisplatin, carboplatin, oxaliplatin eller cyclofosfamid.
Paclitaxel / carboplatin
• Administration af paclitaxel (225 mg / m2) og carboplatin (AUC = 6) med sorafenib (≤ 400 mg to gange dagligt), med en 3-dages afbrydelse af administrationen af sorafenib (de foregående to dage og dagen for administration af paclitaxel / carboplatin ), havde ingen signifikant effekt på paclitaxels farmakokinetik.
• Samtidig administration af paclitaxel (225 mg / m2, en gang hver 3. uge) og carboplatin (AUC = 6) med sorafenib (400 mg to gange dagligt, uden afbrydelse af sorafenib dosering) resulterede i en stigning på 47% i sorafenibs eksponering, en 29% stigning i paclitaxel-eksponering og en 50% stigning i 6-OH paclitaxel-eksponering Farmakokinetikken for carboplatin blev ikke påvirket.
Disse data indikerer, at der ikke er behov for dosisjustering, når paclitaxel og carboplatin administreres samtidigt med sorafenib med en 3-dages afbrydelse af administrationen af sorafenib (de to dage før og dagen for paclitaxel / carboplatin-administration). Bemærk den kliniske relevans af øget eksponering for sorafenib og paclitaxel kort tid efter samtidig administration af sorafenib uden doseringsafbrydelse.
Capecitabin
Samtidig administration af capecitabin (750-1050 mg / m2 to gange dagligt, dag 1-14 hver 21. dag) og sorafenib (200 eller 400 mg to gange dagligt uden doseringsafbrydelse) resulterede ikke i signifikante ændringer i eksponeringen for sorafenib, men en 15 -50% stigning i capecitabin-eksponering og en 0-52% stigning i 5-FU-eksponering Den kliniske relevans af disse små, beskedne stigninger i eksponering for 5-FU er ukendt. Capecitabin og 5-FU ved samtidig administration med sorafenib.
Doxorubicin / Irinotecan
Samtidig behandling med sorafenib resulterede i en stigning på 21% i AUC for doxorubicin. Ved administration med irinotecan, hvis metabolit SN-38 efterfølgende metaboliseres via UGT1A1-vejen, var der en stigning på 67-120% i "AUC for SN-38 og 26 - 42% i AUC for irinotecan. " Den kliniske relevans af disse data er ukendt (se pkt. 4.4).
Docetaxel
Docetaxel (en dosis på 75 eller 100 mg / m2 hver 21. dag) administreret samtidigt med sorafenib (200 mg eller 400 mg to gange dagligt på dag 2 til 19 i et 21-dages behandlingsforløb med en afbrydelse på 3 dage svarende til docetaxel administration) resulterede i en stigning i docetaxel AUC og Cmax på henholdsvis 36 - 80% og 16 - 32%. Forsigtighed anbefales ved samtidig administration af sorafenib og docetaxel (se pkt. 4.4).
Forening med andre agenter
Neomycin
Kombinationen med neomycin, et ikke-systemisk antimikrobielt middel, der bruges til at udrydde den gastrointestinale flora, forstyrrer den enterohepatiske recirkulation af sorafenib (se afsnit 5.2, Biotransformation og metabolisme), hvilket resulterer i nedsat eksponering for sorafenib. Hos raske frivillige behandlet med neomycin i 5 dage faldt den gennemsnitlige sorafenib -eksponering med 54%. Virkningerne af andre antibiotika er ikke undersøgt, men vil sandsynligvis afhænge af deres evne til at forstyrre mikroorganismer med glucuronidaseaktivitet.
04.6 Graviditet og amning
Graviditet
Der er ingen data om brugen af sorafenib hos gravide Dyrestudier har vist reproduktionstoksicitet, herunder misdannelser (se pkt. 5.3) Sorafenib og dets metabolitter har vist sig at passere placenta hos rotter, og sorafenib forventes at forårsage skadelige virkninger Sorafenib bør ikke anvendes under graviditet, medmindre det er klart nødvendigt, og kun efter grundig overvejelse af moderens behov og risikoen for fosteret.
Kvinder i den fertile alder bør anvende effektiv prævention under behandlingen.
Fodringstid
Det vides ikke, om sorafenib udskilles i modermælk. Hos dyr udskilles sorafenib og / eller dets metabolitter i mælk. Da sorafenib kan forringe væksten og udviklingen af den nyfødte (se pkt. 5.3), bør kvinder afbryde amningen under behandling med sorafenib.
Fertilitet
Dyrestudier viser, at sorafenib kan forringe fertiliteten hos mænd og kvinder (se pkt. 5.3).
04.7 Virkninger på evnen til at føre motorkøretøj og betjene maskiner
Der er ikke udført undersøgelser af evnen til at føre motorkøretøj eller betjene maskiner. Der er ingen grund til at tro, at sorafenib påvirker evnen til at føre motorkøretøj eller betjene maskiner.
04.8 Bivirkninger
De vigtigste alvorlige bivirkninger var myokardiskæmi og infarkt, gastrointestinal perforering, lægemiddelhepatitis, blødning og hypertension eller hypertensiv krise.
De mest almindelige bivirkninger var diarré, asteni, alopeci, infektion, hånd-fod hudreaktion (svarer i MedDRA til "palmar-plantar erythrodysaesthesia syndrom") og udslæt.
Bivirkninger rapporteret i forskellige kliniske forsøg eller efter markedsføring er anført i tabel 1, sorteret efter MedDRA og frekvens.Frekvenser er defineret som følger: meget almindelig (≥1 / 10), almindelig (≥1 / 100,
Inden for hver frekvensklasse præsenteres bivirkninger i faldende sværhedsgrad.
Tabel 1: Samlede bivirkninger rapporteret hos patienter i forskellige kliniske undersøgelser eller efter markedsføring.
* Bivirkninger kan være livstruende eller dødelige. Disse hændelser er enten ualmindelige eller mindre hyppige end ualmindelige.
** Hånd-fod hudreaktion svarer til palmar-plantar erythrodysaesthesia syndrom i MedDRA
Lær mere om nogle bivirkninger
Congestiv hjertesvigt
i en virksomhedsstøttet klinisk undersøgelse blev kongestiv hjertesvigt rapporteret som en bivirkning hos 1,9% af patienterne behandlet med sorafenib (N = 2276). I studie 11213 (RCC) blev der rapporteret om bivirkninger i overensstemmelse med kongestiv hjertesvigt hos 1,7% af sorafenib-behandlede patienter og 0,7% af placebo-behandlede patienter. I studie 100554 (HCC) blev sådanne hændelser rapporteret hos 0,99% af sorafenib-behandlede patienter og 1,1% af placebo-behandlede patienter.
Yderligere oplysninger til særlige populationer
I kliniske undersøgelser forekom visse uønskede lægemiddelreaktioner, såsom hånd-fod hudreaktion, diarré, alopeci, vægttab, hypertension, hypokalcæmi og keratoacanthoma / pladecelle hudkarcinom, med en betydeligt højere frekvens hos patienter med differentieret skjoldbruskkirtelkræft sammenlignet med patienter inkluderet i nyre- eller hepatocellulære cellekarcinomundersøgelser.
Ændringer i laboratorietest hos patienter med HCC (studie 3) og RCC (studie 1)
En stigning i lipase og amylase er blevet rapporteret meget almindeligt.En grad 3 eller 4 stigning i lipase Fælles toksicitetskriterierAdvers Events (CTCAE) forekom hos 11% og 9% af patienterne i sorafenib -gruppen i henholdsvis studie 1 (RCC) og undersøgelse 3 (HCC) mod 7% og 9% af patienterne i Sorafenib -gruppen. Behandlet med placebo. En CTCAE grad 3 eller 4 amylaseforhøjelse forekom hos 1% og 2% af patienterne i sorafenib -gruppen i henholdsvis studie 1 og undersøgelse 3 mod 3% af patienterne i begge placebogrupper. Klinisk pancreatitis blev rapporteret hos 2 ud af 451 patienter behandlet med sorafenib (CTCAE grad 4) i studie 1, hos 1 ud af 297 patienter behandlet med sorafenib (CTCAE grad 2) i studie 3 og hos 1 ud af 451 patienter (CTCAE grad 2) behandlet med placebo i studie 1.
Hypophosphatæmi er et meget almindeligt laboratoriefund og blev observeret hos henholdsvis 45% og 35% af sorafenib-behandlede patienter i henholdsvis studie 1 og studie 3 mod 12% og 11% af placebobehandlede patienter. CTCAE grad 3 hypophosphatæmi (1-2 mg / dL) forekom i studie 1 hos 13% af sorafenib-behandlede patienter og hos 3% af placebo-behandlede patienter, mens det i studie 3 forekom hos 11% af patienterne behandlet med sorafenib og hos 2% af patienterne behandlet med placebo Der er ikke rapporteret tilfælde af CTCAE grad 4 hypophosphatæmi (ætiologi af hypophosphatæmi forbundet med sorafenib er ukendt.
CTCAE grad 3 eller 4 laboratorieabnormiteter, herunder lymfopeni og neutropeni, blev observeret hos ≥ 5% af patienterne behandlet med sorafenib.
Hypokalcæmi blev observeret hos 12% og 26,5% af sorafenibbehandlede patienter sammenlignet med 7,5% og 14,8% af patienterne i placebogruppen i henholdsvis studie 1 og studie 3. Tilfælde af hypocalcæmi var milde (CTCAE grad 1 og 2). A CTCAE grad 3 hypokalcæmi (6,0 - 7,0 mg / dL) forekom hos 1,1% og 1,8% af patienterne behandlet med sorafenib og hos 0,2% og 1,1% af patienterne i placebogruppen og en CTCAE grad 4 hypokalcæmi (
I undersøgelser 1 og 3 blev der observeret en reduktion i kalium hos henholdsvis 5,4% og 9,5% af patienterne, der blev behandlet med sorafenib, sammenlignet med 0,7% og 5,9% af patienterne, der fik placebo. De fleste tilfælde af hypokaliæmi var milde (CTCAE grad 1). I disse undersøgelser forekom CTCAE grad 3 hypokaliæmi hos 1,1% og 0,4% af sorafenib-behandlede patienter og 0,2% og 0,7% af patienterne i placebogruppen. Tilfælde af CTCAE grad 4 hypokaliæmi.
Ændringer i laboratorietest hos patienter med DTC (undersøgelse 5)
Hypokalcæmi blev observeret hos 35,7% af sorafenib-behandlede patienter sammenlignet med 11,0% af patienterne i placebogruppen. De fleste tilfælde af hypokalcæmi var lette i sværhedsgrad. CTCAE grad 3 hypokalcæmi forekom hos 6,8% af sorafenib-behandlede patienter og 1,9% af patienterne i placebogruppen, mens CTCAE grad 4 hypokalcæmi forekom hos 3,4% af patienterne. Patienter behandlet med sorafenib og hos 1,0% af patienterne i placebogruppen.
Andre klinisk relevante laboratorieforandringer observeret i studie 5 er vist i tabel 2.
Tabel 2: Laboratorieabnormiteter, der fremkommer ved behandling, rapporteret hos DTC-patienter (studie 5) i den dobbeltblinde fase
* Fælles terminologi -kriterier for bivirkninger (CTCAE), version 3.0
** Etiologien for sorafenib -associeret hypophosphatæmi er ukendt.
Indberetning af formodede bivirkninger
Rapportering af formodede bivirkninger, der opstår efter godkendelse af lægemidlet, er vigtig, da det muliggør løbende overvågning af lægemidlets nytte / risiko -forhold.Professionelle sundhedspersonale anmodes om at rapportere alle formodede bivirkninger via Det Italienske Lægemiddelagentur, websted : www.agenziafarmaco.gov.it/it/responsabili.
04.9 Overdosering
Der er ingen specifikke behandlinger i tilfælde af overdosering af sorafenib. Den højeste dosis sorafenib, der er undersøgt klinisk, er 800 mg to gange dagligt. Bivirkninger observeret efter denne dosering var hovedsageligt diarré og dermatologiske reaktioner. Hvis der er mistanke om overdosering, skal sorafenib seponeres og om nødvendigt initieres understøttende behandling.
05.0 FARMAKOLOGISKE EGENSKABER
05.1 Farmakodynamiske egenskaber
Farmakoterapeutisk gruppe: antineoplastiske midler, proteinkinasehæmmere.
ATC -kode: L01XE05.
Sorafenib er en kinasehæmmer, der har vist både anti-proliferative og anti-angiogene egenskaber in vitro og in vivo.
Virkningsmekanisme og farmakodynamisk virkning
Sorafenib er en kinasehæmmer, der hæmmer spredning af kræftceller in vitro. Sorafenib hæmmer væksten af et bredt spektrum af humane tumorer transplanteret til athymiske mus, hvilket også resulterer i en reduktion i tumorangiogenese.Sorafenib hæmmer aktiviteten af mål til stede i tumorcellen (CRAF, BRAF, V600E BRAF, c-KIT og FLT- 3) og i tumorens blodkar (CRAF, VEGFR-2, VEGFR-3 og PDGFR-Ã). RAF-kinaser er serin / threoninkinaser, mens c-KIT, FLT-3, VEGFR-2, VEGFR-3 og PDGFR-ß er receptortyrosinkinaser.
Klinisk effekt
Sorafenibs sikkerhed og kliniske effekt er blevet undersøgt hos patienter med hepatocellulært karcinom (hepatocellulært carcinomHCC) hos patienter med fremskreden nyrecellekarcinom (nyrecellekræft, RCC) og hos patienter med differentieret kræft i skjoldbruskkirtlen (differentieret thyreoideacancer, DTC).
Hepatocarcinom
Studie 3 (studie 100554) var et multicenter, randomiseret, dobbeltblindet, placebokontrolleret, internationalt fase III-studie med 602 patienter med hepatocellulær cancer. Baseline demografi og sygdomskarakteristika var sammenlignelige mellem sorafenib- og placebogrupperne med hensyn til Estern Cooperative Oncology Group (ECOG) klassificering (grad 0: 54% versus 54%; grad 1: 38% versus 39%; grad 2: 8% versus 7%), til TNM -klassificeringen (trin I:
Undersøgelsen blev afsluttet, efter at en planlagt midlertidig Total Survival (OS) analyse overskred den foruddefinerede effektgrænse. Denne OS-analyse viste en statistisk signifikant stigning i OS for sorafenib-behandlede patienter sammenlignet med placebo-behandlede patienter (HR: 0,69, p = 0,00058, se tabel 3).
I denne undersøgelse er data om patienter med Child Pugh B nedsat leverfunktion begrænset, og kun én Child Pugh C -patient blev inkluderet.
Tabel 3: Effektresultater fra undersøgelse 3 (undersøgelse 100554) i hepatocarcinom
CI = konfidensinterval, HR = Hazard ratio (sorafenib i forhold til placebo)
* statistisk signifikant, da p-værdien var under standard O "cutoff-grænsen, sat til 0,0077
** uafhængig radiologisk gennemgang
En anden fase III, international, multicenter, randomiseret, dobbeltblind, placebokontrolleret undersøgelse (undersøgelse 4, 11849) vurderede den kliniske fordel ved sorafenib hos 226 patienter med fremskreden leverkræft. Denne undersøgelse, udført i Kina, Korea og Taiwan, bekræftede resultaterne af undersøgelse 3 med hensyn til den gunstige fordel-risiko-profil af sorafenib (HR (OS): 0,68, p = 0,01414).
I de foruddefinerede stratificeringsfaktorer (ECOG -klassificering, tilstedeværelse eller fravær af makroskopisk vaskulær invasion og / eller ekstrahepatisk spredning af tumoren) i undersøgelser 3 og 4 var HR konsekvent til fordel for sorafenib frem for placebo. Undersøgende undergruppeanalyser antydede en mindre udtalt behandlingseffekt hos patienter med fjerne metastaser allerede ved baseline.
Nyrecellekarcinom
Sorafenibs tolerabilitet og effekt ved behandling af fremskreden nyrecellekarcinom (RCC) blev undersøgt i to kliniske undersøgelser:
Studie 1 (studie 11213) var et multicenter, randomiseret, dobbeltblindet, placebokontrolleret fase III-studie med 903 patienter. Kun patienter med rencelle renale tumorer og med lav og medium risikofaktor ifølge MSKCC blev registreret. Det endepunktprimære var den samlede overlevelse (OS, samlet set overlevelse) og progressionsfri overlevelse (PFS, Progression Gratis Overlevelse).
Cirka halvdelen af patienterne havde deres generelle tilstand lig med 0 på ECOG -skalaen, og halvdelen af patienterne tilhørte den prognostiske gruppe med en lav score ifølge MSKCC -klassificeringen.
PFS blev vurderet i henhold til RECIST -kriterier med en blindet uafhængig radiologisk gennemgang. PFS -analyse blev udført på 342 hændelser hos 769 patienter. Median PFS -værdien var 167 dage hos sorafenib -behandlede patienter sammenlignet med 84 dage hos patienter, der fik placebo (HR = 0,44; 95% CI: 0,35 - 0,55; p
En "analyse midlertidig (anden analyse midlertidig) for samlet overlevelse (samlet set overlevelse) blev udført på 367 dødsfald hos 903 patienter. Den nominelle alfa -værdi for denne analyse var 0,0094. Median overlevelse var 19,3 måneder hos sorafenib-behandlede patienter sammenlignet med 15,9 måneder hos patienter randomiseret til placebo (HR = 0,77; 95% CI: 0,63-0,95; p = 0,015). På tidspunktet for analysen skiftede cirka 200 patienter fra placebogruppen til sorafenibgruppen.
Undersøgelse 2 var et fase II -studie med randomiseret seponering af behandlingen hos patienter med metastatisk kræft, inklusive RCC. Patienter med stabil sygdom og sorafenib -behandling blev randomiseret til placebo eller til fortsættelse af sorafenib -behandling. PFS hos patienter med RCC var signifikant større (163 dage) for sorafenib-behandlede patienter end dem, der blev observeret hos patienter, der fik placebo (41 dage) (p = 0,0001, HR = 0,29).
Differentieret thyreoideacancer (DTC)
Undersøgelse 5 (undersøgelse 14295) var et internationalt, multicenter, randomiseret, dobbeltblindet, placebokontrolleret fase III-studie udført på 417 patienter med lokalt fremskreden eller metastatisk radiojod ildfast DTC. Progressionsfri overlevelse (PFS), som bestemt ved blindet uafhængig radiologisk vurdering baseret på RECIST-kriterier, var undersøgelsens primære endepunkt. Sekundære endepunkter omfattede samlet overlevelse (OS), tumorresponsrate og varighed af respons Efter progression kunne patienterne modtage open-label sorafenib.
Patienter blev inkluderet i undersøgelsen, hvis de udviklede sig inden for 14 måneder før tilmelding, og hvis de havde en DTC, der var ildfast for radiojod (radioaktivt jod, RAI). DTC ildfast for RAI blev defineret som tilstedeværelsen af en ikke-jodforstærkende læsion på RAI-scintigrafi eller kumulativ administration af RAI ≥ 22,2 GBq eller progression efter RAI-behandling inden for de foregående 16 måneder. Tilmelding eller efter to behandlinger med RAI udført kl. en maksimal afstand på 16 måneder fra hinanden.
Baseline demografi og patientkarakteristika var velafbalanceret i de to behandlingsgrupper. Metastaser var til stede i lungerne hos 86%, i lymfeknuderne hos 51% og i knoglen hos 27% af patienterne. Den mediane kumulative radiojodaktivitet administreret før tilmelding var cirka 14,8 GBq. De fleste patienter havde papillært carcinom (56,8%) efterfulgt af follikulært carcinom (25,4%) og dårligt differentieret carcinom (9,6%).
Mediantiden til PFS var 10,8 måneder i sorafenib -gruppen sammenlignet med 5,8 måneder i placebogruppen. (HR = 0,587; 95% konfidensinterval (CI): 0,454, 0,758; p ensidig
Virkningen af sorafenib på PFS var konstant uanset geografisk region, alder over eller under 60 år, køn, histologi og tilstedeværelse eller fravær af knoglemetastaser.
I en samlet overlevelsesanalyse udført 9 måneder efter skæringsdatoen for den endelige PFS-analyse var der ingen statistisk signifikant forskel i den samlede overlevelse mellem behandlingsgrupperne (HR 0,884; CI 95 %: 0,633; 1,236, ensidig p- værdi på 0,236). Median OS blev ikke opnået i sorafenib-armen, mens det var 36,5 måneder i placebo-armen. Et hundrede og syvogfyrre patienter (75%) randomiseret til placebo og 61 patienter (30%) randomiseret til sorafenib modtog åben sorafenib.
Den mediane behandlingsvarighed i den dobbeltblinde fase var 46 uger (interval 0,3-135) for patienter, der fik sorafenib og 28 uger (interval 1,7-132) for patienter, der fik placebo.
Der blev ikke observeret fuldstændigt svar (komplet svar, CR) i henhold til RECIST kriterier. Den samlede svarprocent (CR + delvis respons, delvis svar (PR)), bestemt ved uafhængig radiologisk evaluering, var højere i sorafenib -gruppen (24 patienter, 12,2%) sammenlignet med placebogruppen (1 patient, 0,5%), p ensidig
En post-post analyse af undergrupper baseret på maksimal tumorstørrelse viste en behandlingseffekt på PFS til fordel for sorafenib sammenlignet med placebo hos patienter med maksimal tumorlæsionsstørrelse på 1,5 cm eller mere (HR 0,54 (95% CI: 0,41-0,71)) , mens en numerisk lavere effekt blev registreret hos patienter med maksimal tumorlæsionsstørrelse mindre end 1,5 cm (HR 0,87 (95% CI: 0,40 -1,89)).
En post-hoc-analyse baseret på kræftrelaterede symptomer på skjoldbruskkirtlen, der var til stede under baseline-betingelser, viste en behandlingseffekt på PFS til fordel for sorafenib frem for placebo hos både symptomatiske og asymptomatiske patienter. HR-værdi for fri overlevelse var 0,39 (95% CI: 0,21- 0,72) for patienter med symptomer ved baseline og 0,60 (95% CI: 0,45 - 0,81) for patienter uden symptomer ved basale tilstande.
Forlængelse af QT -intervallet
I et klinisk farmakologisk studie blev QT / QTc målt hos 31 patienter ved baseline (forbehandling) og efter behandling. Efter en 28-dages behandlingscyklus på tidspunktet for maksimal sorafenibkoncentration blev QTcB forlænget med 4 ± 19 msek og QTcF med 9 ± 18 msek, sammenlignet med baseline for placebogruppen. Ingen patient viste en QTcB- eller QTcF-værdi> 500 msek under EKG-overvågning efter behandlingen (se pkt. 4.4).
Pædiatrisk population
Det Europæiske Lægemiddelagentur har frafaldet forpligtelsen til at indsende resultaterne af undersøgelser i alle undergrupper af den pædiatriske population for kræft i nyre- og nyrebekken (undtagen nefroblastom, nefroblastomatose, klarcellesarkom, mesoblastisk nefrom, nyremedulært carcinom og rabdoid tumor i nyrerne) og carcinom i leveren og intrahepatisk galdegang (undtagen hepatoblastom) og differentieret thyreoideacancer (se pkt.4.2 for information om pædiatrisk brug).
05.2 "Farmakokinetiske egenskaber
Absorption og distribution
Efter administration af sorafenib tabletter er den gennemsnitlige relative biotilgængelighed 38 - 49% sammenlignet med en oral opløsning. Absolut biotilgængelighed er ukendt. Efter oral administration når sorafenib maksimale plasmaniveauer på cirka 3 timer. Når det gives sammen med et måltid med højt fedtindhold, reduceres absorptionen af sorafenib med ca. 30% sammenlignet med administration i fastende tilstand.
Den gennemsnitlige Cmax og AUC stiger mindre end proportionelt med doser over 400 mg to gange dagligt. Plasmaproteinbinding af sorafenib in vitro er 99,5%.
Gentagen dosering af sorafenib i 7 dage resulterede i 2,5 til 7 gange akkumulering sammenlignet med enkelt administration. Sorafenibs steady state opnås inden for 7 dage, med et forhold mellem middelværdi maksimum og lav plasmakoncentration på mindre end 2.
Ligevægtskoncentrationer af sorafenib administreret med 400 mg to gange dagligt blev bestemt hos patienter med DTC, RCC og HCC. Den højeste gennemsnitlige koncentration blev observeret hos patienter med DTC (cirka det dobbelte af det, der blev observeret hos patienter med RCC og HCC), men variabiliteten var høj for alle tumortyper Årsagen til denne højere koncentration hos DTC -patienter er ukendt.
Biotransformation og eliminering
Eliminationshalveringstiden for sorafenib er cirka 25 til 48 timer. Sorafenib metaboliseres primært i leveren via CYP3A4-medieret oxidativ metabolisme og UGT1A9-medieret glucuronokonjugering. Konjugeret sorafenib kan frigives i mave -tarmkanalen ved hjælp af visse bakteriers glucuronidaseaktivitet, hvilket muliggør reabsorption af den ukonjugerede aktive ingrediens. Kombinationen med neomycin er blevet observeret forstyrre denne proces, hvilket reducerer den gennemsnitlige biotilgængelighed af sorafenib med 54%.
Sorafenib tegner sig for ca. 70 - 85% af analytterne, der cirkulerer i steady -state plasma. Otte metabolitter af sorafenib er blevet identificeret, hvoraf fem er fundet i plasma. Den største metabolit af sorafenib, der cirkulerer i plasma, pyridin-N-oxid, udviser styrkein vitro ligner sorafenibs. Denne metabolit tegner sig for cirka 9 - 16% af analytterne, der cirkulerer ved steady state.
Efter oral administration af en dosis på 100 mg sorafenib, blev 96% af dosis genoprettet inden for 14 dage: 77% i fæces og 19% i urinen som glucuronatmetabolitter. Uændret sorafenib, som repræsenterer 51% af dosis, blev genfundet i fæces, men ikke i urinen, hvilket indikerer, at galdeudskillelse af det umetaboliserede aktive stof kan bidrage til eliminering af sorafenib.
Farmakokinetik i særlige kategorier af patienter
Analyse af demografiske data viste, at der ikke er nogen sammenhæng mellem farmakokinetik og alder (op til 65 år), køn eller kropsvægt.
Pædiatrisk population
Der er ikke udført undersøgelser for at verificere sorafenibs farmakokinetik hos pædiatriske patienter.
Race
Der er ingen klinisk relevante forskelle i farmakokinetik mellem kaukasiske og asiatiske forsøgspersoner.
Nedsat nyrefunktion
I fire kliniske fase I-undersøgelser var sorafenibs eksponering ved steady-state hos patienter med let eller moderat nedsat nyrefunktion den samme som hos patienter med normal nyrefunktion. I et klinisk farmakologisk studie (400 mg enkeltdosis sorafenib) blev der ikke observeret nogen sammenhæng mellem sorafenib -eksponering og nyrefunktion hos personer med normal nyrefunktion eller let, moderat eller svært nedsat nyrefunktion. Der er ingen data tilgængelige om patienter, der har behov for dialyse.
Nedsat leverfunktion
Hos patienter med hepatocellulært carcinom (HCC) og med nedsat leverfunktion vurderet som Child-Pugh A eller B (mild til moderat), var eksponeringsværdier sammenlignelige og inden for det område, der blev observeret hos patienter uden nedsat leverfunktion. Farmakokinetikken for sorafenib hos Child-Pugh A- og B-patienter uden HCC var den samme som hos raske frivillige. Der er ingen data for patienter med alvorlig (Child-Pugh C) leverinsufficiens. Sorafenib elimineres primært via leveren, og eksponeringen kan øges i denne patientpopulation.
05.3 Prækliniske sikkerhedsdata
Den prækliniske sikkerhedsprofil af sorafenib blev evalueret hos mus, rotter, hunde og kaniner.
Toksicitetsundersøgelser ved gentagne doser har afsløret ændringer i forskellige organer (degeneration og regenerering) ved eksponeringer under dem, der blev anvendt i kliniske undersøgelser (baseret på en sammenligning af AUC).
Efter gentagen dosering hos unge og voksende hunde blev der observeret effekter på knogler og tænder ved eksponeringer under dem, der blev anvendt i de kliniske undersøgelser. Disse virkninger bestod af ujævn fortykkelse af lårbens vækstplade, medullær hypoplasi i nærheden af de ændrede vækstplader og ændringer i dentins sammensætning. Lignende virkninger blev ikke fremkaldt hos den voksne hund.
Standardprogrammet for genotoksicitetsundersøgelser blev udført, og der blev opnået positive resultater, da en stigning i kromosomale strukturelle aberrationer blev noteret i et assay. in vitro i pattedyrsceller (kinesiske hamster æggestokke) til måling af clastogenicitet i nærvær af metabolisk aktivering. Sorafenib var ikke genotoksisk i Ames -testen eller mikronukleustesten in vivo i musen. Et mellemprodukt i produktionsprocessen, som også er til stede i det endelige aktive stof (in vitro på bakterieceller (Ames -test).Derudover inkluderede batchen af sorafenib, der blev testet i det standard genotoksiske batteri, 0,34% PAPE.
Der er ikke udført kræftfremkaldende undersøgelser med sorafenib.
Der er ikke udført specifikke dyreforsøg med sorafenib for at evaluere effekten på fertilitet. En negativ indvirkning på fertiliteten hos mænd og kvinder må dog forventes, da dyreforsøg med gentagne doser har vist ændringer i mandlige og kvindelige reproduktive organer ved eksponeringer under dem, der blev anvendt i kliniske forsøg (baseret på AUC). Ændringerne bestod typisk af tegn på degeneration og forsinket udvikling af testikler, epididymis, prostata og sædblærer hos rotter. Hunrotter viste central nekrose af corpora lutei og blokering af follikulær udvikling i æggestokkene. Hunde viste tubulær degeneration i æggestokkene. testikler og oligospermi.
Sorafenib har vist sig at være embryotoksisk og teratogent, når det administreres til rotter og kaniner ved eksponeringer under dem, der er anvendt i kliniske undersøgelser. De observerede virkninger omfattede et fald i maternel og føtal kropsvægt, en stigning i antallet af fosterresorptioner og et øget antal eksterne og viscerale misdannelser.
Miljørisikovurderingsundersøgelser har vist, at sorafenibtosylat er potentielt persistent, bioakkumulerende og toksisk for miljøet. Information om miljørisikovurderingen findes i European Public Assessment Report (EPAR) for dette lægemiddel (se afsnit 6.6).
06.0 LÆGEMIDDELOPLYSNINGER
06.1 Hjælpestoffer
Kernen i tabletten:
Croscarmellosenatrium
Mikrokrystallinsk cellulose
Hypromellose
Natriumlaurylsulfat
Magnesiumstearat
Tabletbelægning:
Hypromellose
Macrogol
Titandioxid (E 171)
Rødt jernoxid (E 172)
06.2 Uforenelighed
Ikke relevant.
06.3 Gyldighedsperiode
3 år.
06.4 Særlige opbevaringsforhold
Må ikke opbevares over 25 ° C.
06.5 Den umiddelbare emballages art og emballagens indhold
Karton indeholdende 112 filmovertrukne tabletter (4 x 28) i gennemsigtig blister (PP / aluminium).
06.6 Brugsanvisning og håndtering
Dette lægemiddel kan udgøre en potentiel risiko for miljøet. Ubrugt lægemiddel og affald fra dette lægemiddel skal bortskaffes i overensstemmelse med lokale bestemmelser.
07.0 INDEHAVER AF MARKEDSFØRINGSTILLADELSE
Bayer Pharma AG
13342 Berlin
Tyskland
08.0 MARKEDSFØRINGSTILLADELSESNUMMER
EU/1/06/342/001
037154010
09.0 DATO FOR FØRSTE TILLADELSE ELLER FORNYELSE AF TILLADELSEN
Dato for første godkendelse: 19. juli 2006
Dato for seneste fornyelse: 21. juli 2011
10.0 DATO FOR REVISION AF TEKSTEN
05/2014