Almindelighed
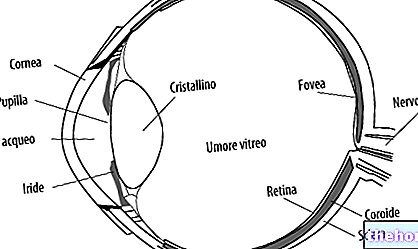
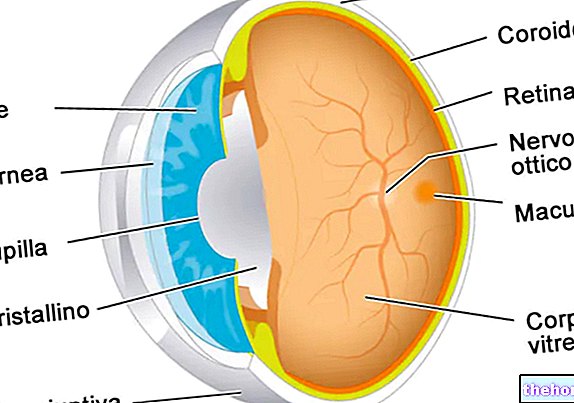
Retinoblastoma (Rb) er en ondartet øjetumor, der udvikler sig fra nethindens celler. Denne kræft kan forekomme i alle aldre, men begyndelsen er mest almindelig i barndommen før fem år.

Barndomskræft er aggressiv: Retinoblastom kan spredes til lymfeknuder, knogler eller knoglemarv. Sjældent involverer det centralnervesystemet (hjerne og rygmarv).
Omkring 90% af børn med retinoblastom har en positiv prognose (sandsynlighed for helbredelse), forudsat at diagnosen er tidlig og behandlingen påbegyndes, inden kræften breder sig. Når det er muligt, er målet med medicinsk intervention at bevare patientens syn.
Årsager
Hændelsesrækken, der fører til tumordebut, er kompleks. Dette begynder, når celler i nethinden udvikler en mutation (eller deletion), der involverer RB1 -tumorsuppressorgenet, der er placeret på q14 -båndet i kromosom 13 (13q14).
Hver celle har normalt to RB1 -gener:
- Hvis mindst en kopi af genet fungerer korrekt, opstår der ikke retinoblastom (men risikoen øges);
- Når begge kopier af genet er muteret eller mangler, sker der ukontrolleret celleproliferation.
I mange tilfælde er det uklart, hvad der præcist inducerer ændringer i RB1 -genet (sporadisk retinoblastom); disse kan skyldes tilfældige genetiske fejl, som f.eks. opstår under reproduktion og celledeling. Det er imidlertid kendt, at de genetiske abnormiteter, der ligger til grund for retinoblastom, også kan overføres fra forældre til børn med et autosomalt dominerende arvsmønster. Det betyder, at hvis en forælder bærer et muteret (dominerende) gen, vil hvert barn have 50% chance for at arve det og 50% chance for at have en normal genetisk sammensætning (recessive gener).
- En lejlighedsvis celle inaktiverer sin eneste normale kopi af RB1 -genet (en kopi er allerede muteret);
- Tabet af de to kopier af RB1 fører til en "overdreven spredning af nethinden.
- En lejlighedsvis celle inaktiverer et af dets normale RB1 -gener;
- Den anden kopi af RB1 -genet er inaktiveret;
- Tabet af de to kopier af RB1 inducerer en overdreven celleproliferation, der fører til retinoblastom.
Genetiske og molekylære egenskaber
- Retinoblastoma var den første tumor, der var direkte forbundet med en "genetisk abnormitet (sletning eller mutation af q14 -båndet i kromosom 13).
- RB1 koder for pRb -proteinet, som spiller en nøglerolle i cellecyklussen: det tillader DNA -replikation og cellecyklusprogression, da det deltager i transkriptionskontrollen af S -fasegener (G1 → † "S).
- Ud over retinoblastom inaktiveres RB1 -genet i blære-, bryst- og lungekræft.
Arveligt retinoblastom
Børn med arveligt retinoblastom har en tendens til at udvikle sygdommen i en tidligere alder end sporadiske tilfælde. Desuden har disse børn en øget risiko for andre ikke-okulære kræftformer, da abnormiteten i RB1-genet er medfødt (dvs. til stede fra fødslen) og påvirker alle celler i kroppen (kendt som en kønsmutation), inklusive dem fra begge. nethinder: Af denne grund har børn med den arvelige form ofte bilateralt retinoblastom frem for kun et øje.
Symptomer
For at lære mere: Symptomer på Retinoblastoma
Det mest almindelige og tydelige tegn på retinoblastom er pupillens unormale udseende, som viser en gråhvid refleksion, når den bliver ramt af en lysstråle (leukocoria eller amaurotisk katterefleks)). Andre tegn og symptomer omfatter: nedsat syn, øjenpine og rødme og forsinkelse i udviklingen. Nogle børn med retinoblastom kan udvikle en skævhed (fejljusterede øjne); i andre tilfælde er det muligt at finde neovaskulært glaukom, som efter et stykke tid kan forårsage forstørrelse af øjet (buftalmo).
Kræftceller kan yderligere invadere øjet og andre strukturer:
- Intraokulært retinoblastom. Retinoblastom kan defineres som intraokulært, når tumoren er helt placeret inde i øjet. Neoplasma kan kun findes i nethinden eller også påvirke andre dele, såsom choroid, ciliary body og en del af synsnerven. Intraokulært retinoblastom spredes derfor ikke til vævene omkring ydersiden af øjet.
- Ekstraokulært retinoblastom. Tumoren kan formeres for at påvirke vævene omkring øjet (orbital retinoblastom). Kræften kan også sprede sig til andre områder af kroppen, såsom hjerne, rygsøjle, knoglemarv og lymfeknuder (metastatisk retinoblastom).
Tilstedeværelsen af orbital forlængelse, uveal involvering og invasion af synsnerven er kendte risikofaktorer for udvikling af metastatisk retinoblastom.
Diagnose
I tilfælde af en positiv familiehistorie undergår patienten regelmæssige øjenundersøgelser for kræftscreening. Hvis medfødt retinoblastom er bilateralt, diagnosticeres det normalt i det første leveår, mens når det kun påvirker det ene øje, kan tilstedeværelsen af tumoren bekræftes i en alder af omkring 18-30 måneder.
Den kliniske diagnose af retinoblastom fastlægges ved undersøgelse af fundus.Tumoren, afhængigt af placeringen, kan være synlig under en simpel undersøgelse af øjet gennem indirekte oftalmoskopi. Billedteknikker kan bruges til at bekræfte diagnosen, definere iscenesættelsen af tumoren (hvor den er, hvor udbredt den er, om den påvirker andre organers funktioner i kroppen osv.) Og afgøre, om behandlingen har været effektiv . Undersøgelser kan omfatte ultralyd, computertomografi (CT) og magnetisk resonansbilleddannelse (MRI).
Den molekylærgenetiske diagnose er mulig ved identifikation af mutationen af RB1-genet. Den cytogenetiske analyse (dvs. af kromosomerne) af perifere blodlymfocytter bruges til at detektere deletioner eller omlejringer, der involverer kromosom 13 (13q14.1-q14. 2) .
Behandlinger
I tilfælde af retinoblastom kan flere behandlingsmuligheder bruges.
Formålet med behandlingen er:
- Eliminer tumoren og red patientens liv;
- Gem øjet, hvis det er muligt;
- Bevar syn så meget som muligt;
- Undgå udvikling af andre kræftformer, som også kan skyldes behandling, især hos børn med arveligt retinoblastom.
Prognose (sandsynlighed for genopretning) og behandlingsmuligheder afhænger af følgende faktorer:
- Stadium af tumoren;
- Patientens alder og generelle sundhedstilstand
- Placering, størrelse og antal tumorfoci;
- Spredning af kræften til andre områder udover øjeæblet
- Hvor sandsynligt er det, at synet kan bevares i det ene eller begge øjne.
De fleste tilfælde af retinoblastom diagnosticeres tidligt og behandles med succes, før kræften kan metastasere uden for øjeæblet, hvilket resulterer i en kur på over 90%.

.jpg)